


Stato gassoso
Definizione

Le differenze macroscopiche tra solidi, liquidi e gas derivano principalmente dai vincoli microscopici che governano l’organizzazione delle particelle costituenti. In particolare, risultano determinanti:
- la distanza media tra le particelle nello specifico stato di aggregazione;
- la natura e l’intensità delle interazioni intermolecolari e interatomiche;
- il grado di ordine spaziale e temporale nella disposizione delle particelle.
Nel solido, la disposizione è altamente ordinata e le particelle sono molto ravvicinate; le forze attrattive sono rilevanti e stabilizzano reticoli cristallini o strutture amorfe con legami localmente cooperativi. La rottura di tale ordine richiede energia considerevole, motivo per cui molte sostanze solide presentano punti di fusione ed ebollizione elevati: l’energia fornita deve vincere le interazioni attrattive e promuovere un aumento dell’entropia strutturale.
Nel gas, al contrario, domina il disordine: la distanza media tra particelle è grande rispetto alle loro dimensioni e le interazioni sono deboli e di breve raggio. Di conseguenza, i punti di fusione ed ebollizione sono tipicamente molto bassi. Un esempio è il neon, che fonde a −248,6 °C ed ebolle a −246,1 °C, valori coerenti con l’esiguità delle forze di dispersione tra atomi monoatomici.
I liquidi occupano una posizione intermedia: la vicinanza tra particelle è paragonabile a quella dei solidi, ma l’ordine a lungo raggio è assente. Le configurazioni microscopiche si riorganizzano in modo continuo, condizionando proprietà caratteristiche come viscosità e tensione superficiale.
Gas quali \( \mathrm{O_2} \) o \( \mathrm{N_2} \) possono essere portati allo stato liquido o solido mediante riduzione della temperatura e/o incremento della pressione, operazioni che favoriscono l’avvicinamento delle particelle e l’insorgenza di interazioni attrattive efficaci. Per esempio, l’azoto liquido si ottiene per distillazione frazionata dell’aria e bolle a circa −195,8 °C alla pressione di 1 atm.
I passaggi di stato sono trasformazioni fisiche: pur variando marcatamente le proprietà fisiche (densità, viscosità, conducibilità termica), l’identità chimica resta invariata. La (Tabella 02.01-01) riassume le principali differenze di proprietà fisiche tra gas, liquidi e solidi.
| Proprietà | Gas | Liquido | Solido |
|---|---|---|---|
| Volume e forma | Si espande fino a riempire il contenitore, assumendone la forma. | Ha un volume proprio; la forma dipende in parte dal contenitore, in parte da massa e temperatura. | Ha volume e forma propri, indipendenti dal contenitore. |
| Densità | Molto bassa (ordine di 10⁻³ g/mL). | Alta (circa 1 g/mL). | Alta (ordine di 1–10 g/mL). |
| Comprimibilità | Elevata. | Molto bassa. | Praticamente nulla. |
| Moto delle particelle | Libero e caotico. | Le particelle scorrono e slittano le une sulle altre. | Le particelle vibrano attorno a posizioni fisse. |
| Distanza intermolecolare | Molto grande. | Ridotta: le particelle sono vicine. | Molto ridotta: le particelle sono strettamente unite. |
| Energia cinetica media | Alta, proporzionale alla temperatura assoluta. | Intermedia. | Bassa: prevalgono le forze attrattive. |
| Esempi biomedici | Gas respiratori (O₂, CO₂, N₂). | Fluidi biologici (sangue, plasma, urina). | Tessuti corporei solidi (ossa, denti). |
Caratteristiche fisiche degli stati della materia
Differenze tra proprietà di solidi, liquidi e gas.
Il gas ideale è un modello di riferimento che collega il comportamento microscopico delle particelle a relazioni macroscospiche tra grandezze di stato. Le variabili fondamentali sono temperatura \( T \), volume \( V \), pressione \( P \) e quantità di sostanza \( n \). Sperimentalmente, variando in modo controllato una di queste grandezze si osservano risposte sistematiche nelle altre, da cui derivano le leggi empiriche dei gas.
L’equazione di stato che sintetizza tali relazioni è:
\( P V = n R T \),
dove \( R \) è la costante dei gas (\( R = 8{,}314\,\mathrm{J\,mol^{-1}\,K^{-1}} \); in unità pratiche: \( R = 0{,}082\,057\,\mathrm{L\,atm\,mol^{-1}\,K^{-1}} \)). Un’illustrazione della legge di Avogadro si ottiene, ad esempio, con una siringa dotata di pistone lubrificato: a \( P \) e \( T \) costanti, raddoppiando la quantità \( n \) di gas introdotto, il volume \( V \) raddoppia in modo proporzionale.
La pressione di un gas nasce dagli urti delle particelle contro le pareti del contenitore ed è definita come forza per unità di superficie. La misura della pressione atmosferica fu resa possibile dal barometro a mercurio ideato da Evangelista Torricelli nel XVII secolo: l’altezza della colonna di mercurio è proporzionale alla pressione esterna (Figura 02.01-01).
Le equivalenze più utilizzate sono:
- \( 1\,\mathrm{atm} = 760\,\mathrm{mm\,Hg} = 760\,\mathrm{torr} \);
- \( 1\,\mathrm{Pa} = 1\,\mathrm{N\,m^{-2}} \), \( 1\,\mathrm{kPa} = 10^3\,\mathrm{Pa} \);
- \( 1\,\mathrm{atm} = 101\,325\,\mathrm{Pa} = 101{,}325\,\mathrm{kPa} \approx 1{,}01325\,\mathrm{bar} \).
Oltre al barometro, per gas contenuti in recipienti si impiegano manometri a U: la differenza di livelli del liquido manometrico fornisce \( \Delta P \), da cui si ricava \( P_{\text{gas}} = P_{\text{est}} \pm \Delta P \) a seconda della configurazione.
Image Gallery

La teoria cinetica molecolare interpreta le leggi dei gas in termini di moto e urti delle particelle. Le assunzioni operative principali sono:
- Un gas è composto da particelle puntiformi (atomi o molecole) in moto rettilineo, casuale e incessante;
- La distanza media tra particelle è molto maggiore del loro diametro, per cui il gas è prevalentemente spazio vuoto;
- Le interazioni attrattive o repulsive tra particelle sono trascurabili tranne che durante le collisioni;
- Le collisioni tra particelle e con le pareti sono perfettamente elastiche, con conservazione dell’energia e della quantità di moto;
- L’energia cinetica media traslazionale è proporzionale alla temperatura assoluta: \( \overline{E_k} = \tfrac{3}{2}k_{\mathrm{B}}T \) per singola particella, e \( \overline{E_k} = \tfrac{3}{2}RT \) per mole di gas.
La distribuzione delle velocità è descritta dalla statistica di Maxwell-Boltzmann; all’aumentare di \( T \), la distribuzione si allarga e si sposta verso velocità maggiori, con aumento della velocità quadratica media \( v_{\mathrm{rms}} = \sqrt{3RT/M} \), dove \( M \) è la massa molare.
La comprimibilità elevata dei gas discende dall’ampio spazio vuoto tra particelle: riducendo il volume, le particelle possono essere avvicinate in modo significativo senza incontrare, entro certi limiti, repulsioni a corto raggio. L’espansione per riempire l’intero contenitore riflette il moto caotico e la mancanza di vincoli coesivi su scala macroscopica.
La densità dei gas è bassa rispetto a liquidi e solidi, in quanto molta parte del volume occupato è priva di materia. Coerentemente, piccole variazioni di \( P \) o \( T \) modificano in modo apprezzabile il volume specifico, come quantificato da \( P V = n R T \).
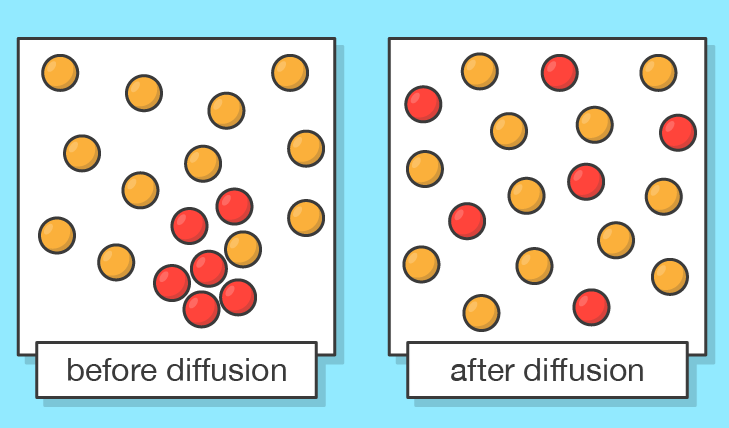
I gas si mescolano e diffondono rapidamente: le particelle, muovendosi in tutte le direzioni, esplorano efficacemente lo spazio disponibile. La dipendenza dalla massa è colta dalla legge di Graham, secondo cui il tasso di diffusione (o effusione) è inversamente proporzionale alla radice quadrata della massa molare: \( r \propto 1/\sqrt{M} \). Molecole leggere diffondono più velocemente di quelle pesanti, in accordo con l’illustrazione in (Figura 02.01-02).
La pressione esercitata su superfici e pareti deriva dagli urti delle particelle: l’incremento del numero di urti al secondo e/o della loro intensità, dovuto a temperature maggiori o volumi minori, accresce \( P \) in modo prevedibile dal modello cinetico.
Le deviazioni dal comportamento ideale sono minime ad alte temperature e basse pressioni, quando le particelle sono ben separate e l’energia cinetica supera le deboli interazioni intermolecolari. All’aumentare della pressione o al diminuire della temperatura, gli effetti di volume proprio e attrazioni (modellabili, ad esempio, con l’equazione di van der Waals) diventano non trascurabili, e il gas reale si discosta dall’ideale.
Image Gallery
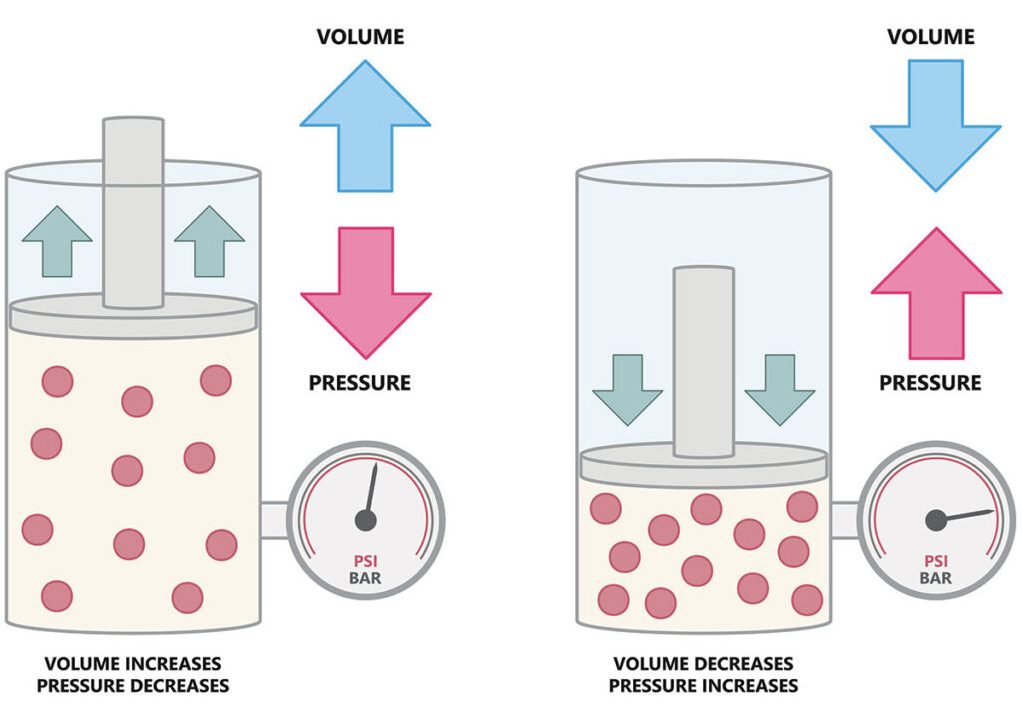
Robert Boyle mostrò sperimentalmente che, mantenendo costanti temperatura e quantità di sostanza, il volume di un gas è inversamente proporzionale alla sua pressione. In termini matematici, il prodotto tra pressione e volume rimane costante lungo una trasformazione isoterma: \( PV = k_1 \). La curva caratteristica è un’iperbole, come rappresentato nella (Figura 02.01-03).
Tra due stati isoterma iniziale e finale vale l’uguaglianza: \[ P_1 V_1 = P_2 V_2. \] Esempio: un campione gassoso occupa \( 8,0 \) L a \( 0,80 \) atm; il prodotto \( PV = 6,4 \) L·atm è costante. Se la pressione viene portata a \( 1,60 \) atm, il volume diventa: \[ (1,60\ \text{atm})\,V_2 = 6,4\ \text{L·atm}\ \Rightarrow\ V_2 = 4,0\ \text{L}. \] Quadruplicando la pressione, il volume si riduce a un quarto, e così via.
Image Gallery
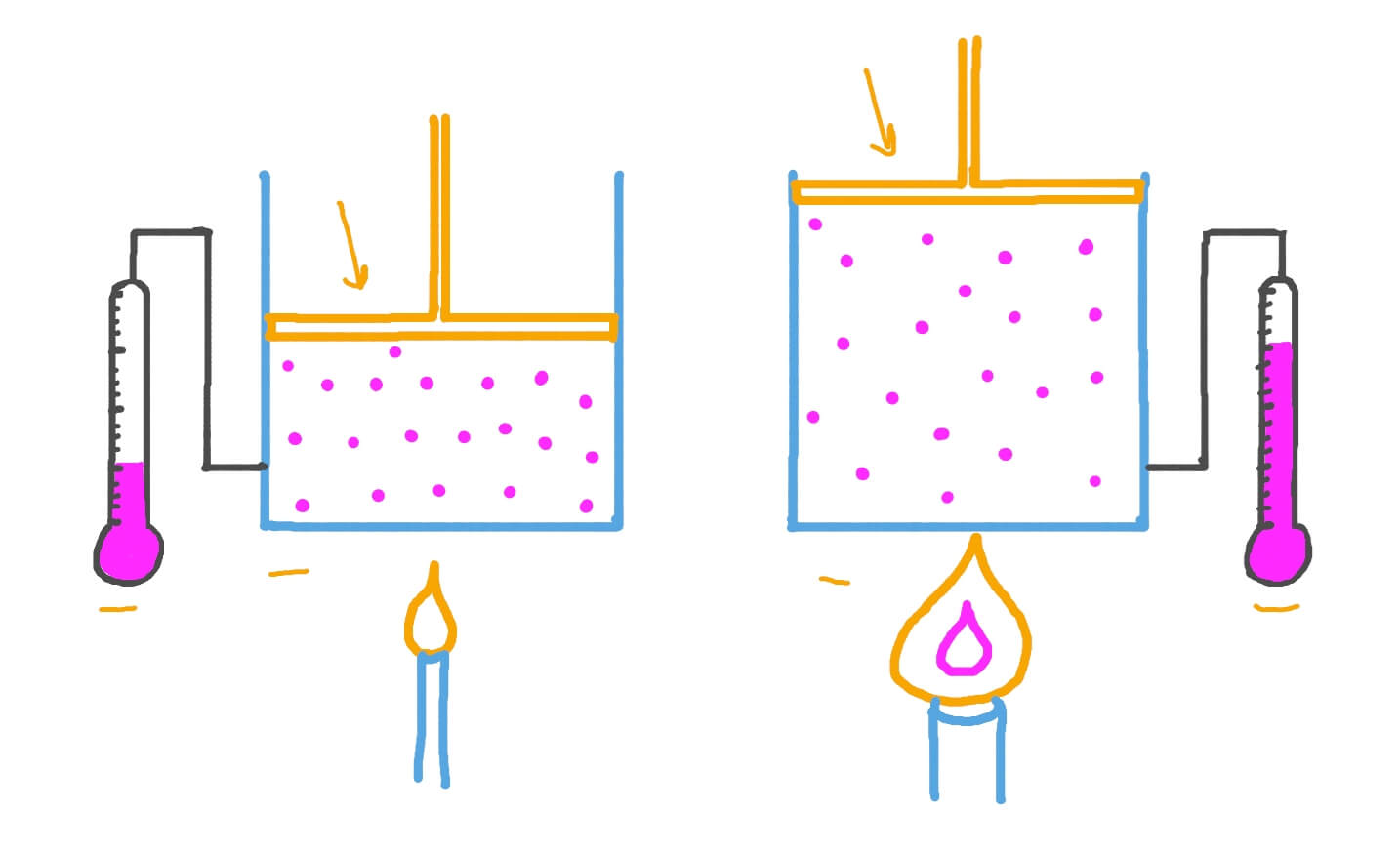
Jacques Charles evidenziò la proporzionalità diretta tra volume e temperatura assoluta di un gas, a pressione e numero di moli costanti. In forma sintetica: \( \frac{V}{T} = k_2 \), con la temperatura misurata in kelvin per garantire linearità. Uguagliando stato iniziale e finale si ottiene \( \frac{V_1}{T_1} = \frac{V_2}{T_2} \).

Esempio: un gas occupa \( 7,50 \) L a \( 300 \) K. Poiché \( V/T \) è costante, se la temperatura aumenta a \( 600 \) K il volume raddoppia a \( 15,0 \) L; a \( 450 \) K il volume risulta \( 11,25 \) L. La dipendenza lineare è mostrata nella (Figura 02.01-04).
Image Gallery
Amedeo Avogadro stabilì che, a temperatura e pressione costanti, il volume di un gas è direttamente proporzionale al numero di moli: \( \frac{V}{n} = k_3 \). Ne consegue che volumi uguali di gas ideali diversi, nelle stesse condizioni di T e P, contengono lo stesso numero di particelle. Scrivendo tra due stati: \( \frac{V_1}{n_1} = \frac{V_2}{n_2} \).
Esempio: se \( 0,50 \) mol di gas occupano \( 12,0 \) L, allora \( 1,00 \) mol, alle medesime T e P, occupano \( 24,0 \) L; \( 1,50 \) mol ne occuperanno \( 36,0 \) L, e così via.
Il volume occupato da una mole di gas si chiama volume molare. In condizioni standard di temperatura e pressione (STP, \( T = 273,15 \) K; \( P = 1 \) atm) il volume molare di un gas ideale vale circa \( 22,4 \) L·mol\(^{-1}\). Pertanto, 1 mol di \( \mathrm{N_2} \), \( \mathrm{O_2} \), \( \mathrm{H_2} \) o He occupa lo stesso volume a STP. In ambito IUPAC è anche diffuso l’uso di \( P = 1 \) bar, per cui il volume molare è circa \( 22,7 \) L·mol\(^{-1}\); la differenza deriva dal diverso valore della pressione standard.
La densità di un gas è definita come \( d = \frac{\text{massa}}{\text{volume}} \). A STP, poiché \( 1 \) mol occupa \( 22,4 \) L, la densità può essere calcolata da \( d = \frac{M}{22,4\ \text{L}} \), dove \( M \) è la massa molare:
- Neon (Ne): massa molare \( M = 20,18 \) g·mol\(^{-1}\); dunque \[ d = \frac{20,18\ \text{g}}{22,4\ \text{L}} \approx 0,901\ \text{g/L}; \];
- Anidride carbonica (\(\mathrm{CO_2}\)): massa molare \( M = 44,01 \) g·mol\(^{-1}\); quindi \[ d = \frac{44,01\ \text{g}}{22,4\ \text{L}} \approx 1,964\ \text{g/L}. \].
Equazione di stato del gas ideale
Le leggi di Boyle, Charles e Avogadro si combinano nell’equazione di stato del gas ideale: \[ PV = nRT, \] dove \( R \) è la costante dei gas ideali. Usando P in atmosfere, V in litri, n in moli e T in kelvin, \( R = 0,082057 \) L·atm·K\(^{-1}\)·mol\(^{-1}\). In unità SI, \( R = 8,314 \) J·mol\(^{-1}\)·K\(^{-1}\):
- Unità coerenti per l’uso di \( R = 0,082057 \) L·atm·K\(^{-1}\)·mol\(^{-1}\): P in atm;
- V in L;
- n in mol;
- T in K.
Esempio: un campione contiene \( 0,25 \) mol di gas a \( 298 \) K e \( 1,00 \) atm. Il volume è \[ V = \frac{nRT}{P} = \frac{0,25 \times 0,082057 \times 298}{1,00} \approx 6,12\ \text{L}. \]
In una miscela, ciascun componente gassoso esercita una pressione come se fosse da solo nel contenitore alla stessa temperatura; la pressione totale è la somma delle pressioni parziali: \[ P_{\text{tot}} = p_1 + p_2 + p_3 + \dots = \sum_i p_i. \] Esempio: in un recipiente si miscelano Ne, \( \mathrm{O_2} \) e \( \mathrm{CO_2} \) con pressioni parziali rispettivamente \( 0,40 \) atm, \( 0,25 \) atm e \( 0,10 \) atm. La pressione totale è \( P_{\text{tot}} = 0,75 \) atm. È spesso utile introdurre la frazione molare \( x_i \), per cui \( p_i = x_i\,P_{\text{tot}} \).
L’atmosfera terrestre realizza un caso naturale di somma di pressioni parziali, in cui numerose specie gassose contribuiscono alla pressione complessiva con valori anche molto piccoli (per esempio, i gas serra come \( \mathrm{CO_2} \) hanno effetti fisici rilevanti pur con pressioni parziali modeste).
Il gas ideale è un modello in cui le particelle non hanno volume proprio e non interagiscono tra loro; nella realtà, deboli attrazioni e repulsioni interparticellari e il volume finito delle molecole producono deviazioni dall’idealità. Tali deviazioni diventano evidenti ad alta pressione (le particelle sono più vicine) e a bassa temperatura (le attrazioni hanno più effetto).
Le molecole polari, con distribuzione di carica non uniforme, manifestano attrazioni elettrostatiche più marcate e si discostano maggiormente dal comportamento ideale rispetto a molecole piccole e non polari. In condizioni blande (pressioni moderate, temperature lontane dalla condensazione) l’equazione dei gas ideali fornisce comunque stime utili.
Per descrivere meglio i gas reali, si impiegano correzioni empiriche, come l’equazione di van der Waals: \[ \left(P + a\frac{n^2}{V^2}\right)\,(V - nb) = nRT, \] dove \( a \) quantifica le attrazioni e \( b \) il volume escluso. Un indicatore sintetico della deviazione è il fattore di comprimibilità \( Z = \frac{PV}{nRT} \): \( Z = 1 \) per un gas ideale, mentre per gas reali \( Z \neq 1 \) e varia con T e P.


