


Scrittura delle strutture di Lewis di molecole e di ioni poliatomici
TOPICS
Definizione

Le formule di Lewis dei singoli atomi, basate sugli elettroni di valenza, costituiscono il punto di partenza per descrivere come gli atomi si organizzano in molecole e ioni poliatomici. Le strutture di Lewis permettono di visualizzare la distribuzione degli elettroni di legame e delle coppie solitarie, offrendo un modello semplice ma efficace per correlare connettività e assetto elettronico. Per redigere in modo coerente una struttura di Lewis è utile seguire un insieme di regole operative, integrandole con controlli sistematici su conteggi elettronici e cariche formali:
- Impostare lo scheletro con i simboli chimici nell’ordine di connettività, affiancando gli atomi che si legano direttamente; quando non noto, occorre proporre una connettività ragionevole in base a linee guida: l’atomo meno elettronegativo tende a occupare il centro; H e F sono quasi sempre terminali; gli alogeni in genere sono terminali; l’ossigeno preferisce posizioni terminali salvo nei perossidi o come ponte; il carbonio forma frequentemente catene e strutture ramificate C–C;
- Determinare il totale degli elettroni di valenza sommando i contributi atomici, \(N_{\text{val,tot}}=\sum_i V_i\), dove \(V_i\) è il numero di elettroni di valenza dell’atomo i-esimo;
- Unire l’atomo centrale con ciascun atomo adiacente mediante legami singoli, ricordando che ogni legame impiega due elettroni; verificare che gli elettroni utilizzati non superino \(N_{\text{val,tot}}\);
- Completare gli ottetti degli atomi terminali con coppie solitarie e, se restano elettroni, assegnarli all’atomo centrale; l’idrogeno soddisfa la sua configurazione con due soli elettroni (duetto); esistono eccezioni alla regola dell’ottetto: specie elettron-deficienti (es. B, Be) e, per elementi dal terzo periodo in poi, possibili espansioni dell’ottetto in presenza di legami multipli o dativi;
- Se qualche atomo (spesso quello centrale) non raggiunge l’ottetto, trasformare coppie solitarie su atomi adiacenti in legami multipli, introducendo legami doppi o tripli ove chimicamente plausibile, fino a soddisfare gli ottetti e mantenere una connettività chimica sensata;
- Effettuare un controllo finale: ricontare gli elettroni e valutare le cariche formali con \( \mathrm{CF} = V - N_{\text{non\,leganti}} - \tfrac{1}{2}N_{\text{di\,legame}} \); sono preferibili strutture con cariche formali ridotte in valore assoluto e con cariche negative su atomi più elettronegativi.
La procedura per gli ioni poliatomici replica quella per le molecole neutre, con una correzione cruciale: il computo degli elettroni di valenza deve includere la carica dello ione. In termini compatti, \[ N_{\text{val,tot}}=\sum_i V_i \pm |q| , \] dove si aggiunge \(|q|\) per anioni e si sottrae \(|q|\) per cationi. Tale correzione riflette l’acquisto o la perdita netta di elettroni associata alla carica complessiva:
- Per cationi poliatomici, sottrarre un elettrone per ogni unità di carica positiva; esempio: per NH4+, \(N(5) + 4\times H(1) - 1 = 8\) elettroni di valenza;
- Per anioni poliatomici, aggiungere un elettrone per ogni unità di carica negativa; esempio: per NO3−, \(N(5) + 3\times O(6) + 1 = 24\) elettroni di valenza.
Nell’atmosfera, H2, O2 e N2 sono specie biatomiche covalenti, ma presentano comportamenti chimici molto differenti: l’idrogeno è altamente infiammabile, l’ossigeno sostiene vigorosamente la combustione, mentre l’azoto molecolare mostra una notevole inerzia chimica e costituisce circa l’80 % dell’aria, fungendo da diluente dell’O2 (circa 20 %). Una parte sostanziale di queste differenze si comprende esaminando ordine di legame, energia e lunghezza di legame.
Le strutture di Lewis di H2, O2 e N2 sono riportate di seguito.
 |
| Molecola di idrogeno – formula strutturale di H₂ |
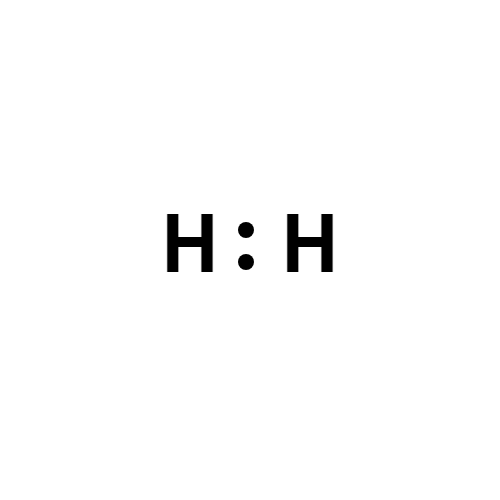 |
| Molecola di idrogeno – struttura di Lewis di H₂ |
Per O2 (12 elettroni di valenza complessivi) la rappresentazione di Lewis che massimizza gli ottetti prevede un legame doppio:
 |
| Molecola di ossigeno – struttura di Lewis di O₂ |
Per N2 (10 elettroni di valenza), la descrizione più semplice comporta un legame triplo tra gli atomi di azoto:
![What is the molecular geometry of a nitrogen molecule \\[{N_2}\\]?](https://cmds-test.vedantu.com/prod/question-sets/a7aac2a7-0c22-489f-87c8-bdfd96b504713104928581183890569.png) |
| Molecola di azoto – struttura di Lewis di N₂ con triplo legame |
| Molecola di azoto – rappresentazione alternativa della struttura di Lewis di N₂ |
Ne segue che:
- N2 possiede un legame triplo, con tre coppie elettroniche condivise;
- O2 presenta tipicamente un legame doppio;
- H2 contiene un singolo legame covalente.
L’energia richiesta per rompere un legame (energia di dissociazione o energia di legame media) è un indicatore della sua robustezza: valori maggiori implicano legami più stabili. In termini generali, a parità di coppia di atomi, l’ordine di legame cresce passando da legame singolo a doppio a triplo, e così fanno anche le energie di legame; le lunghezze di legame, viceversa, decrescono. Per esempi rappresentativi: H–H ≈ 436 kJ/mol; O=O ≈ 499 kJ/mol; N≡N ≈ 941 kJ/mol. La più elevata energia di dissociazione di N≡N spiega la scarsa reattività di N2, mentre H–H e O=O, meno energetici, risultano più facilmente scindibili in processi chimici o radicalici. Si osservi inoltre che lo stato fondamentale di O2 è tripletto con due elettroni spaiati, una caratteristica che contribuisce alla sua particolare reattività e al comportamento paramagnetico, aspetti che la semplice struttura di Lewis non evidenzia esplicitamente.
Talvolta più formule di Lewis, tutte conformi alla regola dell’ottetto e con conteggi elettronici corretti, possono essere scritte per la stessa specie. In tali circostanze la vera distribuzione elettronica è delocalizzata e non può essere descritta da una singola rappresentazione: si ricorre allora al concetto di risonanza e all’ibrido di risonanza, che sintetizza il contributo di tutte le forme ammissibili.
Consideriamo il diossido di zolfo, SO2, con scheletro S–O–O.
 |
| Diossido di zolfo |
Il numero totale di elettroni di valenza è \[ N_{\text{val,tot}} = V(\mathrm{S}) + 2\,V(\mathrm{O}) = 6 + 2\times 6 = 18. \] Partendo da legami singoli S–O e completando gli ottetti sugli ossigeni, lo zolfo risulta elettron-deficiente; per soddisfare gli ottetti si introducono legami multipli, ottenendo due forme limite equivalenti, nelle quali il doppio legame S=O è localizzato alternativamente su un ossigeno o sull’altro:
- una forma con S=O a sinistra e S–O a destra con cariche formali appropriate;
- una forma con S–O a sinistra e S=O a destra, simmetrica alla precedente.
Le corrispondenti strutture sono:
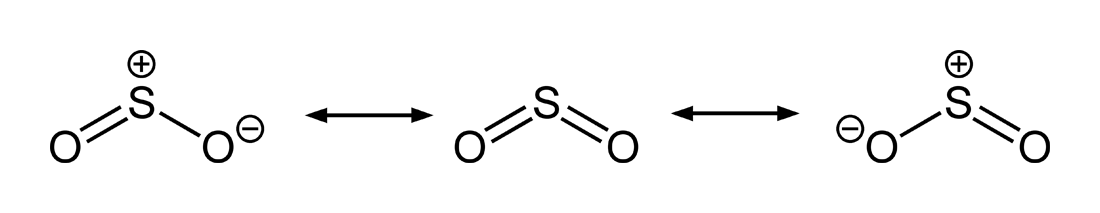 |
| Molecola di acido carbonico – struttura di Lewis di H₂CO₃ |
Le misure sperimentali indicano che i due legami S–O sono equivalenti e hanno lunghezza intermedia tra un singolo e un doppio legame; ciò implica un ordine di legame medio di circa 1,5 per ciascun legame S–O. La descrizione più fedele è quindi l’ibrido di risonanza, nel quale la densità elettronica π è delocalizzata su entrambi i legami S–O:
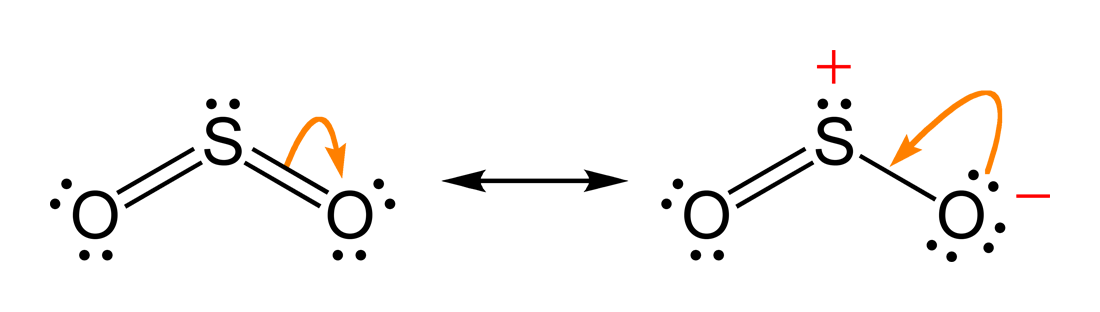 |
| Momento dipolare – distribuzione di carica e polarità del legame covalente |
La risonanza non rappresenta un’alternanza temporale tra forme, ma una delocalizzazione statica degli elettroni: l’ibrido possiede energia inferiore a qualunque singola forma limite, riflettendo una stabilizzazione per risonanza. Quando sono disponibili più strutture contributrici equivalenti, la stabilizzazione è generalmente maggiore. Nella scelta delle forme di risonanza più rilevanti si considerano criteri qualitativi:
- minimizzazione del numero e del valore assoluto delle cariche formali;
- posizionamento di cariche negative su atomi più elettronegativi e cariche positive su atomi meno elettronegativi;
- massimizzazione del numero di ottetti completi, compatibilmente con le eccezioni note;
- coerenza con dati sperimentali quali lunghezze di legame e distribuzioni di carica.
Il quadro risultante consente di collegare struttura elettronica e proprietà: legami equivalenti, lunghezze intermedie e reattività si comprendono in termini di delocalizzazione, un concetto chiave tanto nella chimica inorganica quanto in quella organica e dei materiali π-coniugati.
La regola dell’ottetto costituisce un’utile approssimazione per interpretare il legame covalente e l’assetto elettronico di numerose specie molecolari, ma non è universale. Si incontrano tre tipologie ricorrenti di eccezioni:
- molecole ed ioni a ottetto incompleto, tipiche di elementi poveri di elettroni come berillio, boro e alluminio, che tendono a stabilizzarsi con 4 o 6 elettroni nel guscio di valenza anziché 8;
- radicali con numero dispari di elettroni di valenza, come il monossido di azoto NO, per i quali risulta intrinsecamente impossibile accoppiare tutti gli elettroni in coppie;
- specie ipervalenti di elementi del terzo periodo e dei successivi, capaci di ospitare più di otto elettroni attorno all’atomo centrale, generando “gusci di valenza espansi” (esempi classici includono PF5 e SF6).
Queste deviazioni non invalidano il modello di Lewis, ma ne delimitano il dominio di applicazione, suggerendo quando considerare stabilizzazioni alternative (legami a tre centri, delocalizzazione, accettazione di coppie elettroniche, ecc.). I casi illustrati di seguito offrono esempi didattici delle principali eccezioni.
La morfologia molecolare influenza in modo cruciale proprietà fisiche, polarità, spettroscopia e reattività. La combinazione di strutture di Lewis e della teoria VSEPR (Valence-Shell Electron-Pair Repulsion) consente una previsione razionale delle geometrie: le regioni di densità elettronica attorno all’atomo centrale (coppie di legame e coppie solitarie) si dispongono massimizzando la distanza reciproca per minimizzare le repulsioni elettrostatiche. Nel legame covalente, la condivisione direzionale di elettroni tra nuclei conferisce orientazioni spaziali definite; al contrario, l’interazione elettrostatica nei solidi ionici è essenzialmente non direzionale. L’assetto risultante delle coppie elettroniche determina l’architettura della molecola.
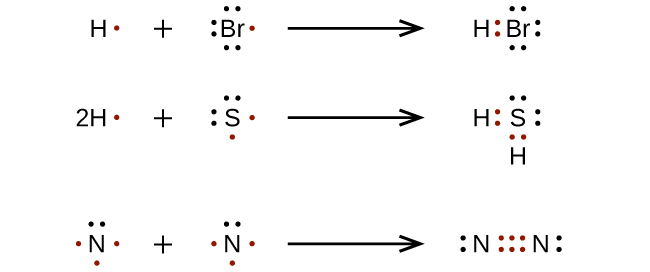 |
| Formazione di legami covalenti – esempi di molecole HBr, H₂S e N₂ |
In VSEPR i legami multipli sono trattati come un’unica regione elettronica; la geometria dipende dunque dal numero totale di regioni attorno al centro, mentre la presenza di coppie solitarie comprime gli angoli di legame rispetto ai valori ideali. Il principio guida è: repulsioni coppia solitaria–coppia solitaria > coppia solitaria–coppia di legame > coppia di legame–coppia di legame. Applichiamo questi criteri ai sistemi seguenti, di cui si considerano le strutture di Lewis.
BeH2
Il berillio in BeH2 è circondato da due atomi legati e non possiede coppie solitarie. Le due regioni elettroniche si dispongono ai capi opposti dello spazio, generando una geometria lineare con angolo H–Be–H pari a 180° (Figura 01.16-01). Altri esempi lineari includono BeF2, CS2, CO2 e HCN. Nel cianuro di idrogeno, la struttura di Lewis è H–C≡N:, e gli atomi legati al carbonio si collocano a 180°. Ai fini predittivi, è più efficace contare gli atomi legati (o domini elettronici) intorno al centro piuttosto che il numero di elettroni di legame.
BF3
Nel trifluoruro di boro tre atomi sono legati al boro e non si hanno coppie solitarie sul centro. La configurazione che minimizza le repulsioni dispone le tre regioni elettroniche in un piano ai vertici di un triangolo, con geometria trigonale planare e angoli F–B–F di 120° (Figura 01.16-02):
 |
| Molecola di trifluoruro di boro – struttura di Lewis di BF₃ |
Specie isoelettroniche o dello stesso gruppo periodico mostrano spesso geometrie analoghe; ad esempio, composti di alluminio(III), quali AlH3 in fase gassosa, presentano tendenza alla planarità trigonale. È importante ricordare che BF3 è elettron-deficiente e agisce da acido di Lewis, potendo accettare una coppia elettronica per formare addotti che colmano l’ottetto.
CH4
Il metano possiede quattro atomi legati al carbonio e nessuna coppia solitaria sul centro. Le quattro regioni elettroniche si collocano ai vertici di un tetraedro con angoli H–C–H pari a 109,5° (Figura 01.16-03). Comportamenti paralleli si riscontrano in SiCl4 e SiH4, dove il silicio, appartenente allo stesso gruppo del carbonio, conduce a strutture tetraedriche.
NH3
Nell’ammoniaca sono presenti tre coppie di legame e una coppia solitaria sull’azoto. L’impalcatura elettronica sottostante resta di tipo tetraedrico, ma la coppia solitaria esercita repulsione maggiore delle coppie di legame, comprimendo gli angoli e producendo una forma trigonale piramidale con angolo H–N–H di circa 107° (Figura 01.16-04). La deviazione rispetto ai 109,5° tetraedrici è coerente con l’ordine di repulsione sopra indicato.
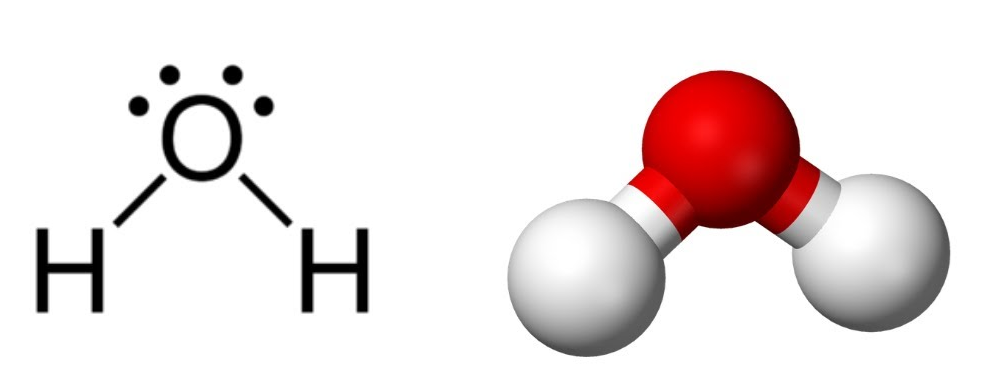
H2O
Nella molecola d’acqua l’ossigeno ospita due coppie di legame e due coppie solitarie. La disposizione elettronica resta prossima alla tetraedrica, ma la presenza di due coppie solitarie intensifica la compressione degli angoli, determinando una struttura piegata (angolare) con angolo H–O–H di circa 104,5° (Figura 01.16-05). Le principali geometrie elementari — lineare, trigonale planare, trigonale piramidale, angolare e tetraedrica — sono sintetizzate nella (Tabella 01.16-01). Per centri con cinque o sei regioni elettroniche si ottengono, rispettivamente, la bipiramide trigonale (PF5) e l’ottaedro (SF6).
Relazioni strutturali periodiche
Gli esempi considerati coinvolgono come atomi centrali elementi dei gruppi 2, 13, 14, 15 e 16 (storicamente IIA, IIIA, IVA, VA e VIA). È spesso utile, sebbene non infallibile, assumere che composti con lo stesso elemento centrale condividano la medesima geometria VSEPR. Analogamente, elementi dello stesso gruppo mostrano tendenze strutturali affini grazie a configurazioni elettroniche di valenza analoghe.
Nel gruppo dell’ossigeno (gruppo 16) O, S e Se possiedono sei elettroni di valenza e formano con l’idrogeno H2O, H2S e H2Se. Poiché l’acqua è angolare, ci si attende — e si osserva — che anche H2S e H2Se siano angolari, con strutture di Lewis analoghe. Scendendo lungo il gruppo, l’aumento delle dimensioni atomiche e la diversa efficacia delle coppie solitarie nel comprimere gli angoli comportano variazioni sistematiche negli angoli H–E–H (E = O, S, Se), generalmente più piccoli per l’acqua e leggermente più ampi per gli idruri più pesanti.
 |
| Molecola d’acqua – struttura di Lewis di H₂O |
segue che anche H2S e H2Se saranno molecole angolari con strutture di Lewis simili, ossia:

|
| Molecola di solfuro di idrogeno – struttura di Lewis di H₂S |
| Molecola di seleniuro di idrogeno – struttura di Lewis di H₂Se |
Questa corrispondenza periodica si estende a molti altri gruppi rappresentativi, pur con eccezioni dovute a effetti sterici, differenze di elettronegatività e delocalizzazione elettronica.
Molecole più complesse
In molecole policentriche la geometria complessiva risulta dalla combinazione delle geometrie locali attorno a ciascun centro. Nel dimetil etere, CH3–O–CH3, coesistono tre atomi centrali: l’ossigeno e i due carboni dei gruppi metile. Ciascun carbonio del gruppo CH3 presenta una disposizione tetraedrica simile a quella del metano:
 |
| Confronto tra carbocatione CH₃⁺ e carbanione CH₃⁻ |
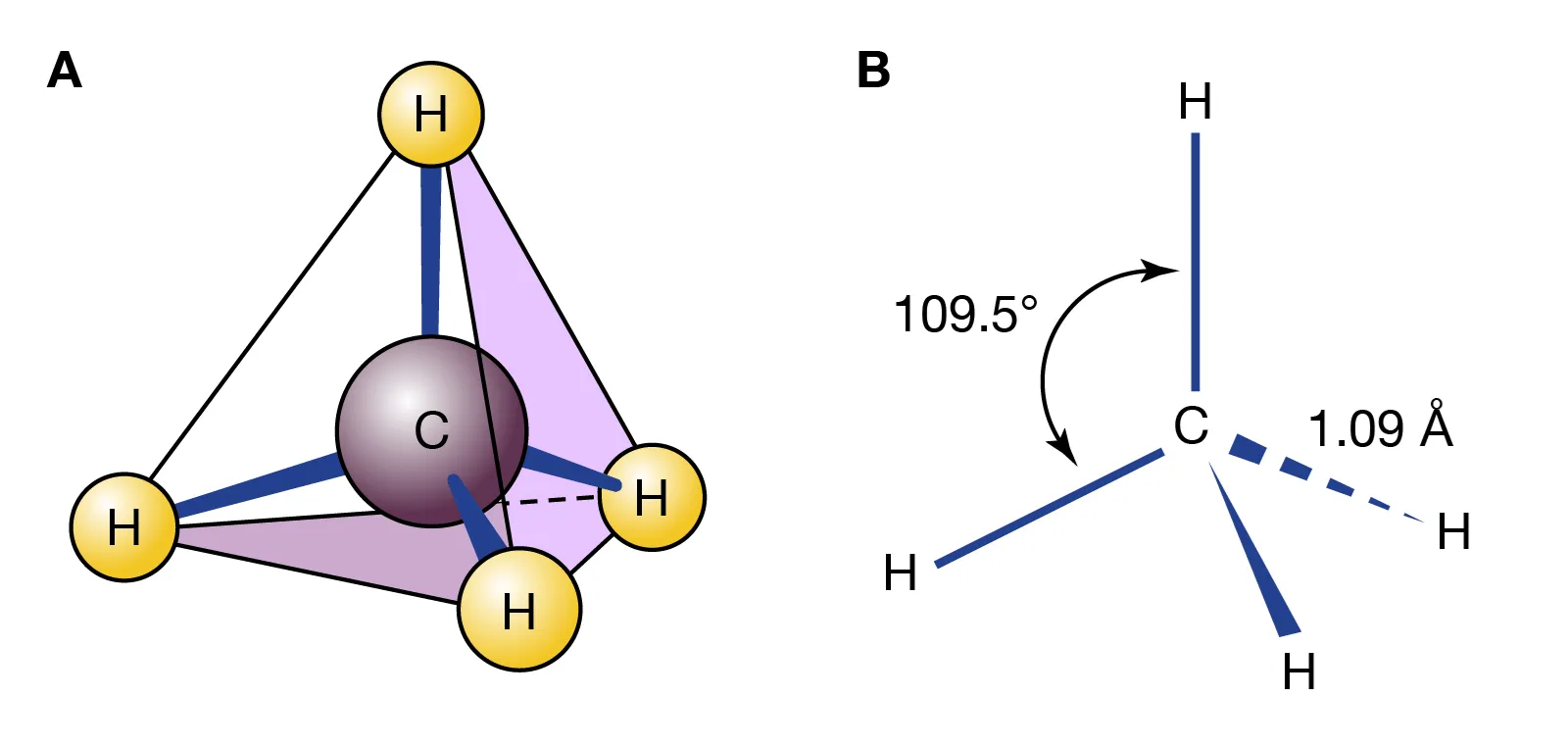 |
| Geometria tetraedrica del metano – angoli e lunghezze di legame in CH₄ |
L’ossigeno, che condivide due coppie di legame e possiede due coppie solitarie, conferisce al segmento O–C una geometria locale analoga a quella dell’acqua con un angolo C–O–C di circa 104°–105°, come indicato nella (Figura 01.16-06). Questa descrizione risulta coerente con la polarità e le proprietà fisiche del dimetil etere.
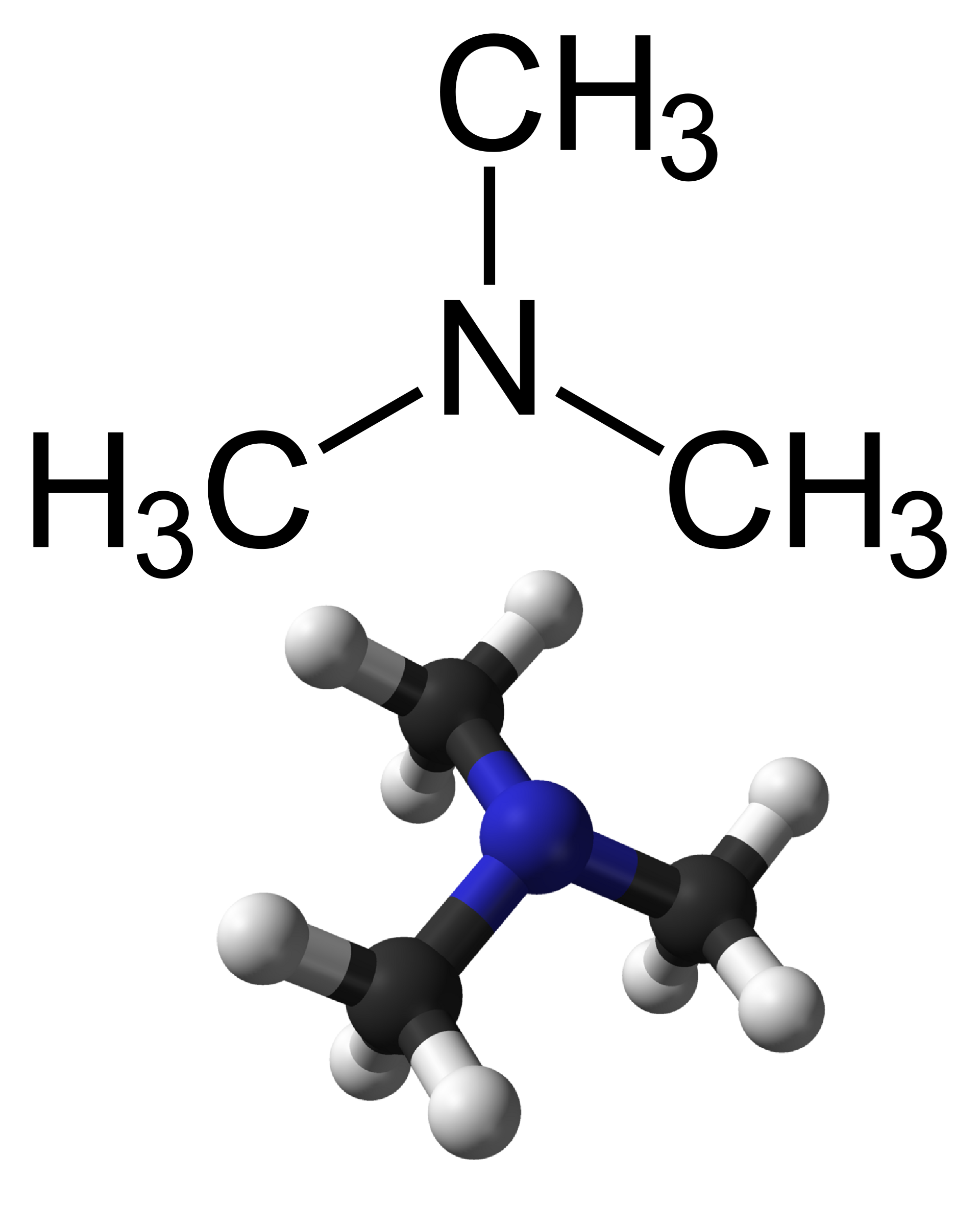
La trimetilammina, (CH3)3N, appartiene alla famiglia delle ammine. I carboni dei gruppi metile mantengono geometria tetraedrica; l’azoto è circondato da tre legami e una coppia solitaria, generando una forma trigonale piramidale, strettamente correlata a quella dell’ammoniaca, come nella (Figura 01.16-07). Gli angoli C–N–C risultano prossimi a 107°, con lievi variazioni dovute all’effetto induttivo dei sostituenti alchilici e a interazioni steriche.
Una rappresentazione geometrica accurata, fondata su Lewis e VSEPR, è essenziale per interpretare polarità, momenti di dipolo, interazioni intermolecolari e tendenze reattive; gli stessi modelli forniscono una base per comprendere perché alcune specie rispettino l’ottetto, mentre altre ne devino in modo prevedibile.
Image Gallery
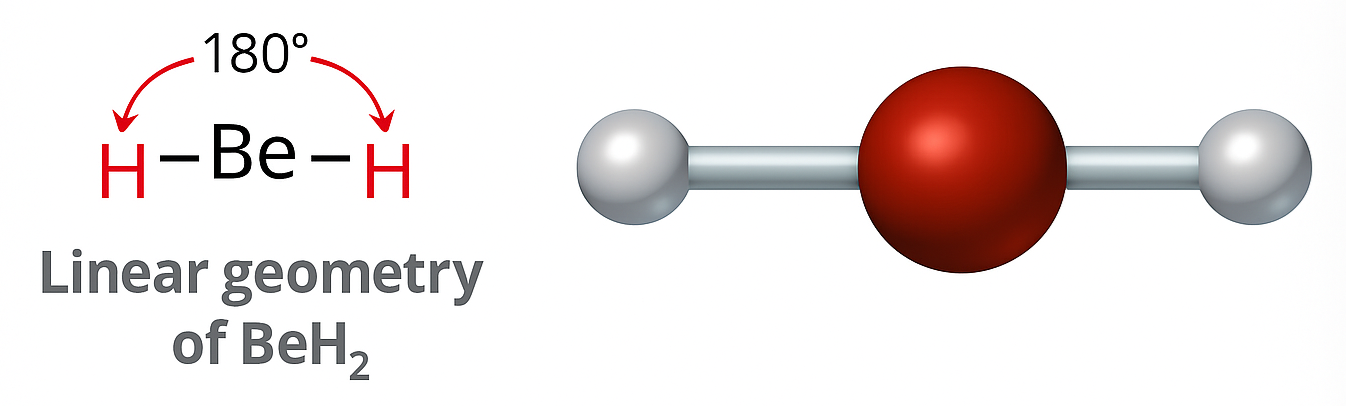
Image Gallery
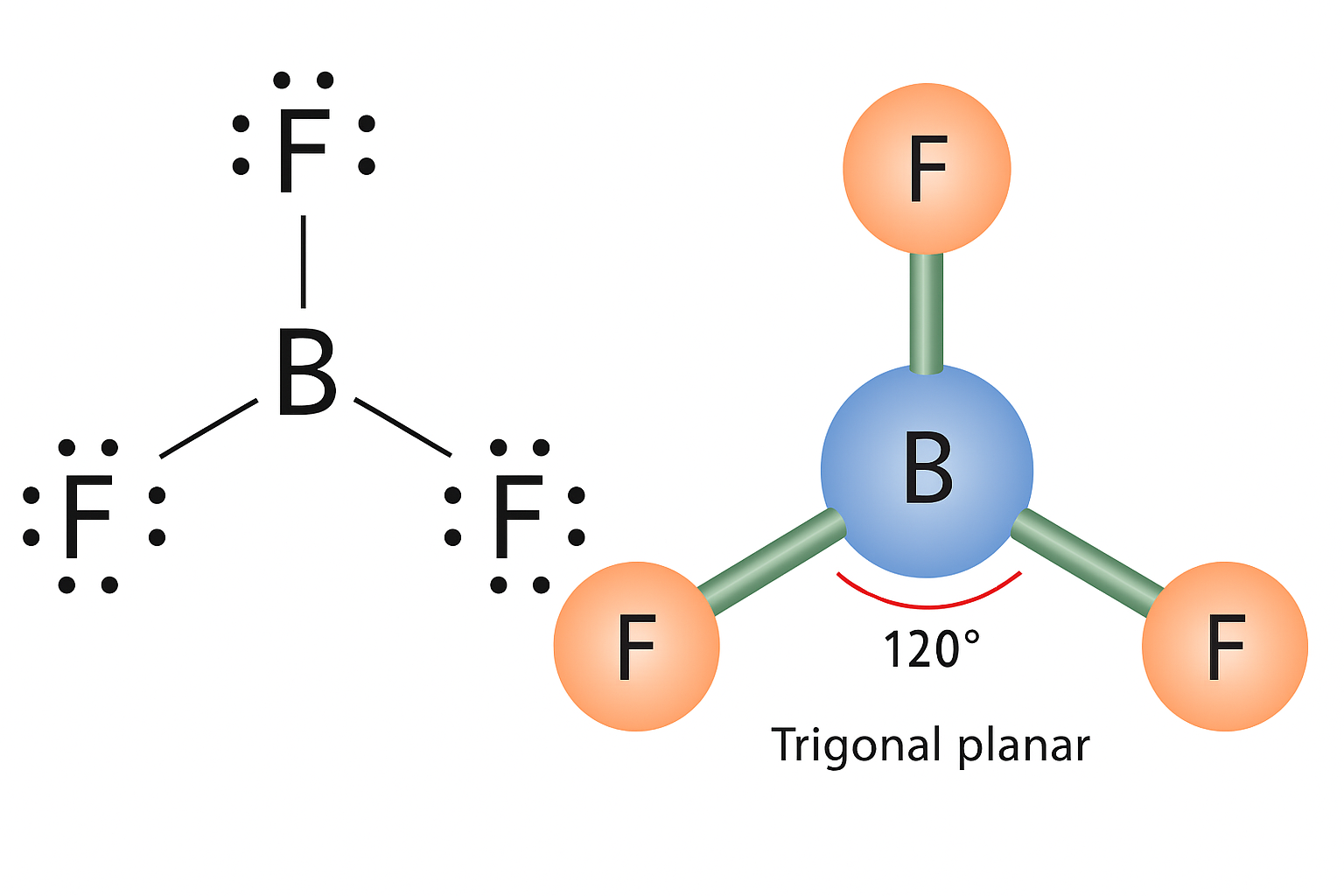
Image Gallery
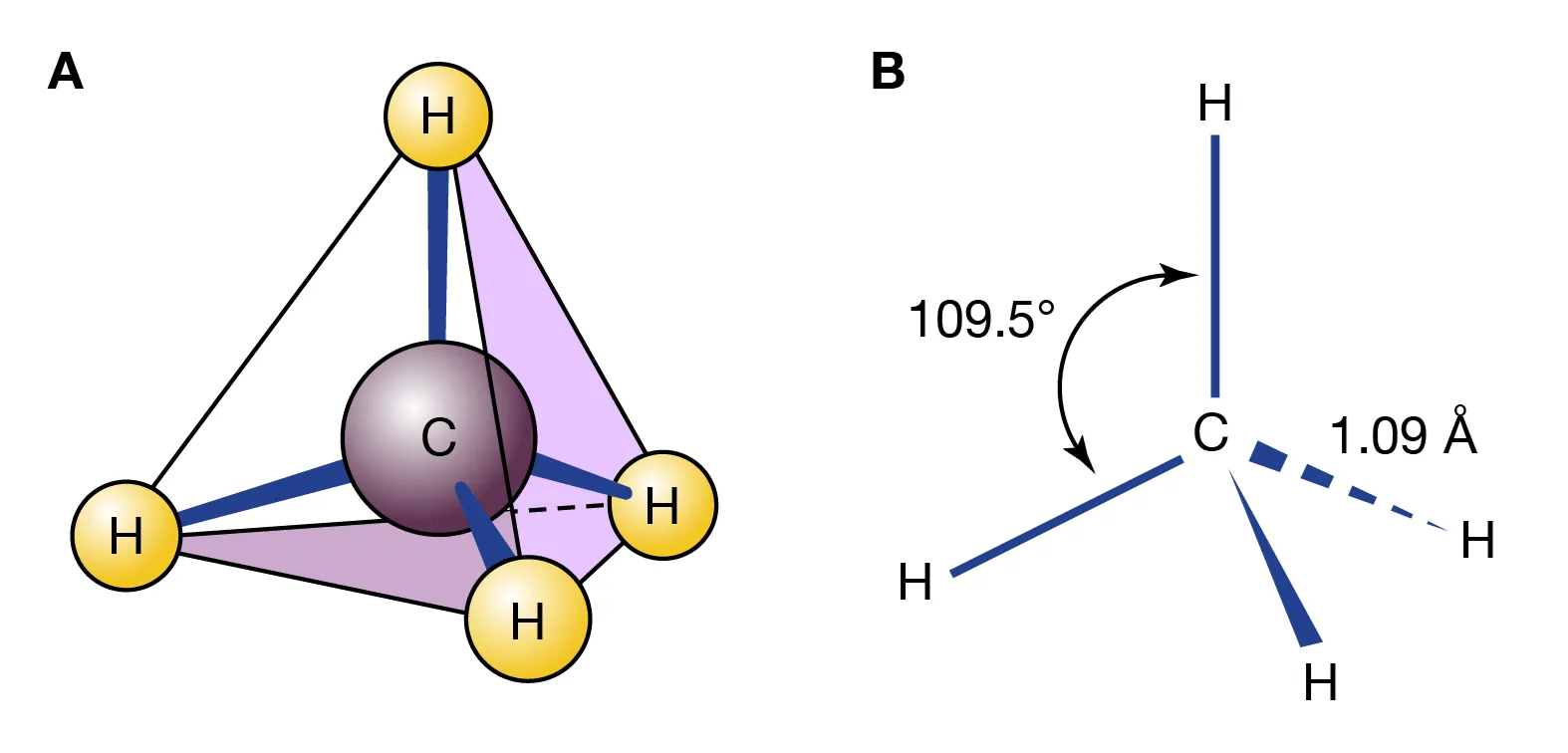
Image Gallery
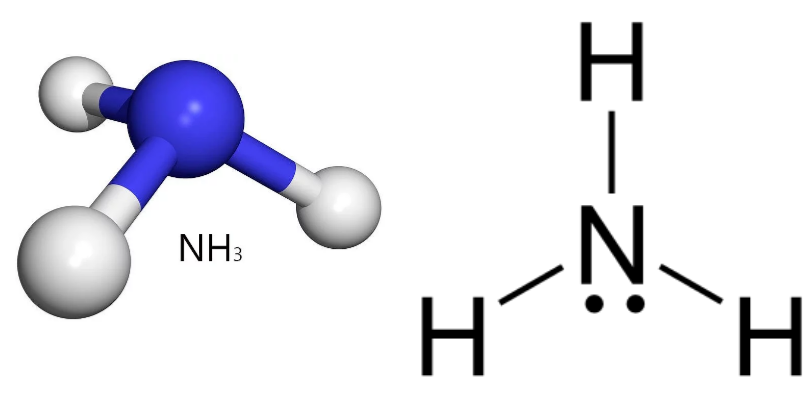
Image Gallery
| Atomi legati | Coppie di elettroni di non-legame | Angolo di legame | Struttura molecolare | Esempio | Nota biochimica |
|---|---|---|---|---|---|
| 2 | 0 | 180° | Lineare | CO₂ | Molecola apolare, fondamentale nella respirazione cellulare |
| 3 | 0 | 120° | Trigonale planare | SO₃ | Struttura tipica di composti solforati ossidati |
| 2 | 1 | <120° | Angolare | SO₂ | Molecola polare, implicata nell’inquinamento atmosferico |
| 4 | 0 | 109,5° | Tetraedrica | CH₄ | Geometria base del carbonio organico (metano, amminoacidi) |
| 3 | 1 | ~107° | Trigonale piramidale | NH₃ | Molecola polare, struttura simile a quella di ammine biologiche |
| 2 | 2 | ~104,5° | Angolare | H₂O | Molecola essenziale, forte polarità → solvente universale |
| 5 | 0 | 90° e 120° | Bipiramidale trigonale | PCl₅ | Presente in composti di fosforo, rilevanza in biochimica energetica |
| 6 | 0 | 90° | Ottaedrica | SF₆ | Geometria di ioni metallici complessi (es. Mg²⁺ in sistemi enzimatici) |
Struttura e forma delle molecole
La disposizione geometrica di una molecola è determinata sia dalle coppie di elettroni non leganti sull’atomo centrale sia dal numero di atomi a esso collegati.
Image Gallery
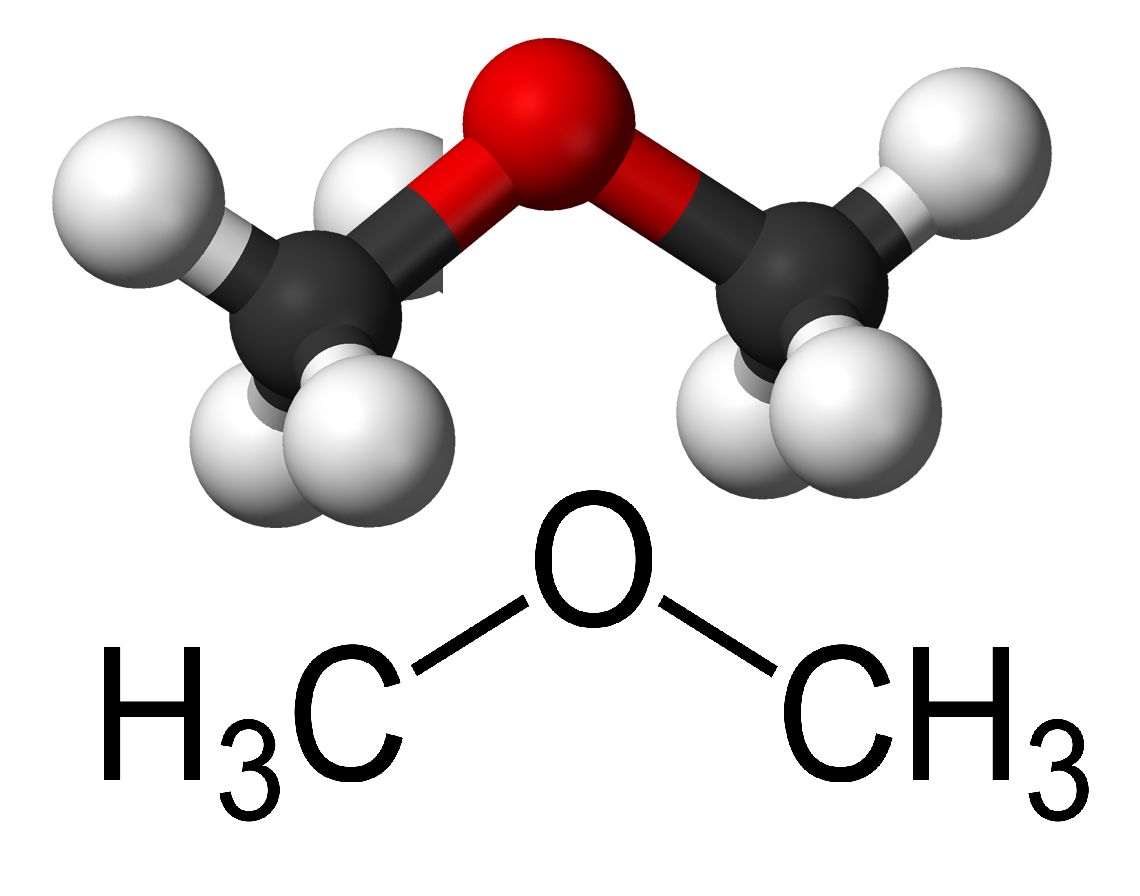
Image Gallery
Una molecola si definisce polare quando il baricentro delle cariche positive non coincide con quello delle cariche negative; in caso contrario è apolare. In presenza di un campo elettrico esterno, i dipoli molecolari tendono ad orientarsi lungo il campo stesso. La polarità può essere descritta quantitativamente tramite il momento di dipolo, una grandezza vettoriale indicata con \( \boldsymbol{\mu} \), pari a \( \boldsymbol{\mu} = q\,\boldsymbol{r} \) per un diatomico ideale (carica separata \(q\) a distanza \(r\)), mentre per molecole poliatomiche si ottiene come somma vettoriale dei contributi dei singoli legami e delle coppie solitarie: \( \boldsymbol{\mu} = \sum_i \boldsymbol{\mu}_i \). L’unità di misura comunemente usata in chimica è il Debye (D).
Il legame covalente è non polare quando gli elettroni di legame sono condivisi in maniera equivalente tra gli atomi coinvolti. Questo accade per diatomiche omonucleari come H2, O2, N2, Cl2 e F2, nelle quali gli atomi hanno la stessa elettronegatività e la densità elettronica è simmetricamente distribuita. La molecola di idrogeno rappresenta il caso più semplice: si può scrivere come H:H oppure H–H, e la densità elettronica media risulta centrata tra i due nuclei positivi; i centri delle cariche coincidono e il dipolo complessivo è nullo.
Quando due atomi con diversa elettronegatività formano un legame, la condivisione degli elettroni diventa diseguale e il legame è polare. Nel fluoruro di idrogeno, HF, l’elevata elettronegatività del fluoro attira la densità elettronica verso di sé: ne deriva un estremo parzialmente negativo sul fluoro e uno parzialmente positivo sull’idrogeno.
 |
| Dipolo elettrico nella molecola di HF – distribuzione della densità elettronica |
La direzione della polarità del legame si indica spesso con una freccia puntata verso l’atomo più elettronegativo; in alternativa si usano le notazioni parziali di carica: \( \delta^- \) per l’atomo arricchito di elettroni e \( \delta^+ \) per quello impoverito.
 |
| Momento dipolare nella molecola d’acqua – polarità dei legami e risultante |
Un legame covalente polare non implica necessariamente una molecola polare: la polarità molecolare dipende sia dall’intensità dei momenti di dipolo dei singoli legami sia dalla loro disposizione geometrica. In altre parole, il vettore risultante \( \boldsymbol{\mu} \) può annullarsi per simmetria, anche quando i legami sono polari. Per stabilire se una specie molecolare sia polare o apolare conviene procedere in modo sistematico a partire dalla struttura di Lewis:
- Tracciare la struttura di Lewis correttamente, identificando eventuali coppie solitarie sull’atomo centrale;
- Determinare la geometria elettronica e molecolare mediante il modello VSEPR;
- Stimare la differenza di elettronegatività tra atomi legati, per individuare i legami polari;
- Eseguire la somma vettoriale dei momenti di dipolo dei legami e delle coppie solitarie: se la risultante è nulla, la molecola è apolare; altrimenti è polare.
Quando l’atomo centrale non possiede coppie solitarie e gli atomi terminali sono tutti equivalenti, la simmetria molecolare tende ad annullare i dipoli di legame. Un esempio classico è l’anidride carbonica, CO2: la struttura è lineare e i due dipoli C=O, uguali e opposti, si elidono, rendendo la molecola apolare. Analogamente, in CCl4 la geometria tetraedrica con quattro legami C–Cl identici produce cancellazione totale dei dipoli.
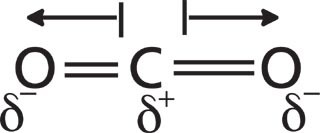 |
| Polarità della molecola di anidride carbonica – distribuzione delle cariche parziali in CO₂ |
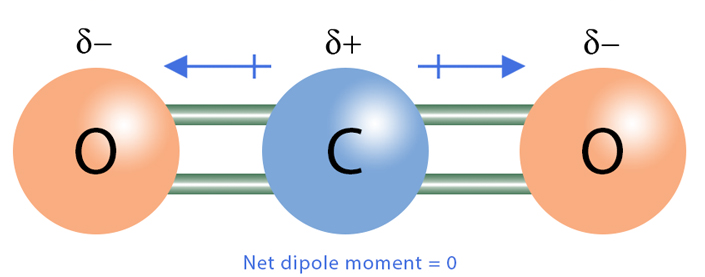 |
| Molecola lineare di CO₂ – dipoli opposti e momento risultante nullo |
La presenza di coppie solitarie sull’atomo centrale altera la simmetria e contribuisce alla risultante dipolare. Molecole con una coppia solitaria sull’atomo centrale sono spesso polari. Nell’ammoniaca, NH3, la geometria molecolare è piramidale trigonale: i tre legami N–H sono polari verso l’azoto e la coppia solitaria “spinge” la geometria fuori dal piano, generando un momento di dipolo non nullo.
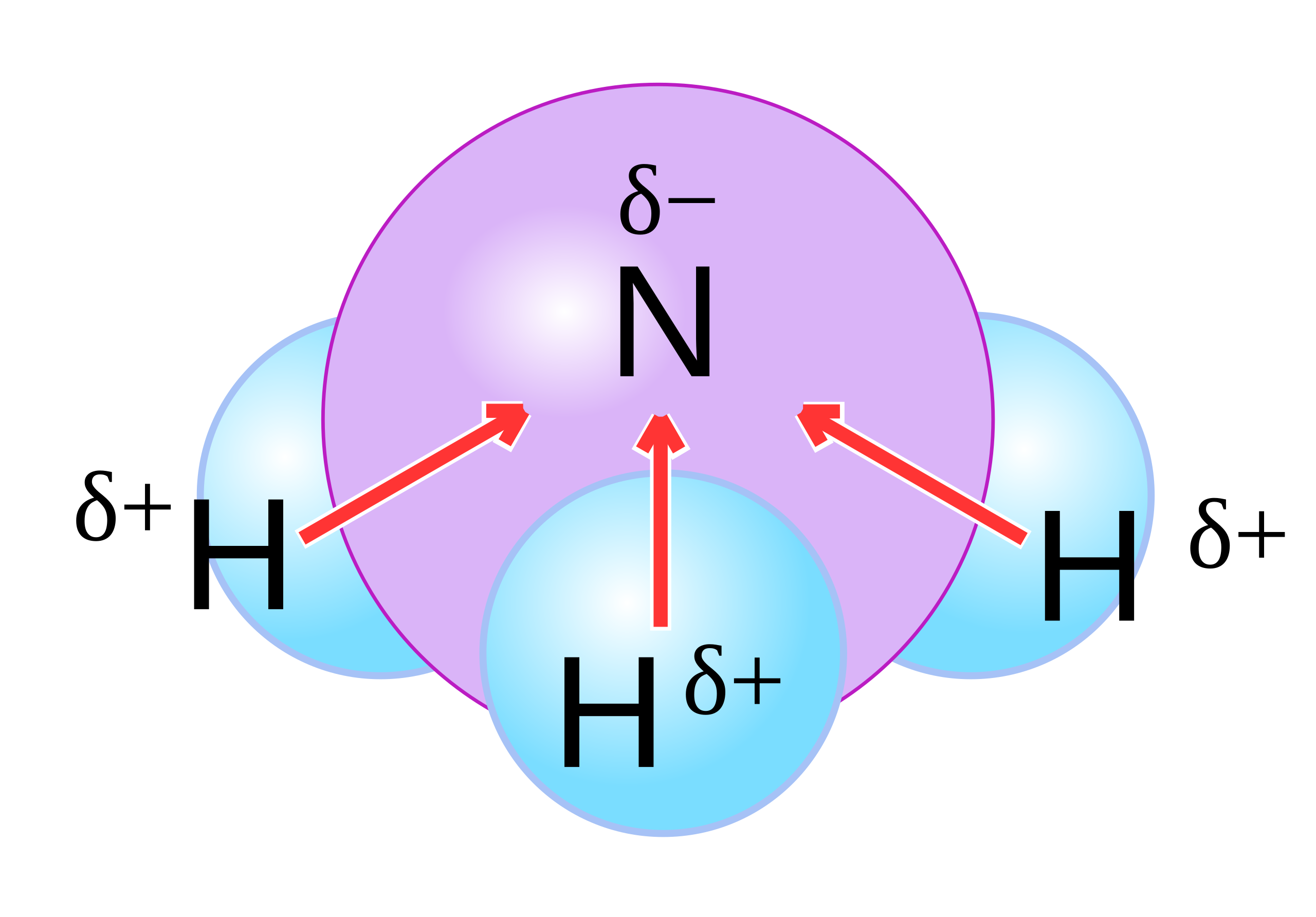 |
| Polarità della molecola di ammoniaca – distribuzione delle cariche parziali in NH₃ |
Con più coppie solitarie sul centro, la polarità è di norma presente, sebbene esistano eccezioni legate alla simmetria complessiva. L’acqua, H2O, possiede due coppie solitarie sull’ossigeno e una geometria angolare (circa 104,5°), che impedisce l’annullamento dei due momenti O–H: il risultato è una molecola fortemente polare con il polo negativo localizzato verso l’ossigeno.
 |
| Legami a idrogeno tra molecole d’acqua – interazioni intermolecolari in H₂O |
La distribuzione di carica è spostata verso l’ossigeno.
Ulteriori esempi consolidano il criterio geometrico. Il trifluoruro di boro, BF3, pur contenendo tre legami B–F polari, è apolare perché planare trigonale con dipoli disposti a 120°, la cui somma vettoriale è nulla. Al contrario, il cloroformio, CHCl3, risulta polare: sebbene la geometria sia tetraedrica, la presenza di atomi terminali non equivalenti (H e Cl) impedisce l’annullamento completo dei dipoli. Ciò dimostra che sia la simmetria sia l’omogeneità degli atomi terminali sono determinanti.
Un’utile analogia è quella della composizione di forze: ogni legame polare e ogni coppia solitaria contribuiscono con un vettore “tiro” nella direzione dell’atomo più elettronegativo (o della regione di densità elettronica libera). Se i “tiri” risultano bilanciati nello spazio, la molecola è apolare; se rimane una risultante, la molecola è polare. La conoscenza della polarità molecolare consente di prevedere proprietà macroscopiche quali solubilità, punto di fusione ed ebollizione, e l’intensità delle interazioni intermolecolari (per esempio, dipolo–dipolo o legame a idrogeno).
Il modello VSEPR, fondato sulle strutture di Lewis, è uno strumento efficace per prevedere la disposizione geometrica degli atomi in una molecola. La rappresentazione di Lewis, però, interpreta il legame covalente come mera condivisione di una coppia elettronica e, in tal modo, descrive nello stesso modo il legame in H2 e in F2. Le misure spettroscopiche e termochimiche mostrano invece differenze nette nelle energie e nelle lunghezze di legame di queste molecole, incongrue con una trattazione puramente lewisiana. Per spiegare tali osservazioni occorrono modelli quantomeccanici più articolati: in particolare, la teoria del legame di valenza (Valence Bond, VB) e la teoria degli orbitali molecolari (Molecular Orbitals, MO). In questa sede faremo riferimento soprattutto alla teoria VB, richiamando quando opportuno le implicazioni geometriche ed energetiche del modello.
Nel quadro VB, un legame covalente si stabilisce quando due orbitali atomici su atomi adiacenti si sovrappongono e ospitano una coppia elettronica con massima densità di probabilità nella regione compresa tra i nuclei. Da questo postulato conseguono tre idee cardine, cui se ne aggiunge una quarta per le molecole poliatomiche:
- Accoppiamento di spin nella regione di sovrapposizione. In conformità con il principio di esclusione di Pauli, l’orbita di legame risulta occupabile al più da due elettroni, che devono avere spin opposti. Per H2, la sovrapposizione dei due orbitali 1s porta a una coppia con spin antiparalleli localizzata tra i nuclei (Figura 01.16-08);
- Ottimizzazione della sovrapposizione e stabilità del legame. L’energia di legame cresce con l’incremento della sovrapposizione degli orbitali coinvolti; la qualità della sovrapposizione dipende dalla forma e dall’orientazione degli orbitali. La sovrapposizione frontale genera un legame di tipo \(\sigma\), con densità elettronica massima lungo l’asse internucleare; esempi, illustrati in (Figura 01.16-08), includono H2 (s–s), HF (s–p) e F2 (p–p). In generale, tutti i legami singoli derivanti da sovrapposizione frontale di orbitali s, p o ibridi sono legami \(\sigma\);
- Sovrapposizione laterale e legami multipli. La sovrapposizione fianco a fianco di orbitali p genera un legame \(\pi\), più debole di un \(\sigma\) in quanto caratterizzato da minore sovrapposizione effettiva. In doppi e tripli legami uno è \(\sigma\), gli altri sono \(\pi\). In N2 (Figura 01.16-09) il triplo legame è costituito da un \(\sigma\) (sovrapposizione 2px–2px) e due \(\pi\) (2py–2py e 2pz–2pz), ciascuno con due lobi di densità elettronica separati da un piano nodale contenente l’asse internucleare;
- Ibridizzazione degli orbitali atomici. Per molecole poliatomiche, la semplice sovrapposizione di orbitali s e p degli atomi isolati non è sufficiente a giustificare le geometrie osservate. Il mescolamento lineare di orbitali atomici di valenza sullo stesso centro, proposto da Pauling, produce orbitali ibridi con orientazioni spaziali coerenti con gli angoli di legame sperimentali.
Un caso paradigmatico è il metano, CH4. La promozione elettronica del carbonio genera quattro elettroni spaiati collocati in 2s, 2px, 2py e 2pz, come schematizzato in seguito:
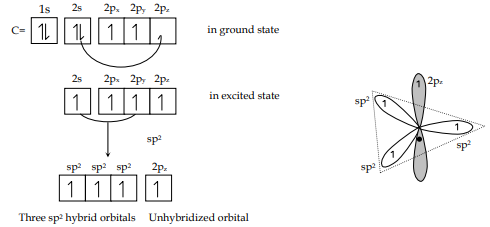 |
| Ibridazione sp² del carbonio – formazione di tre orbitali ibridi e uno p non ibridato |
Se si mantenessero orbitali atomici puri, i tre p sono ortogonali con angoli di 90° e il 2s è sferico: non si potrebbero giustificare quattro legami equivalenti e angoli H–C–H pari a 109,5°. Il ricorso all’ibridizzazione consente di ottenere quattro orbitale equivalenti orientati tetraedricamente, coerenti con le misure sperimentali. In termini generali:
- L’ibridizzazione è un costrutto teorico utile a razionalizzare la geometria del legame in molecole poliatomiche;
- È definita come combinazione lineare di almeno due orbitali atomici non equivalenti sullo stesso atomo, producendo orbitali ibridi con morfologia e direzionalità nuove, determinate dal set di orbitali mescolati;
- Il numero di orbitali ibridi generati coincide con il numero di orbitali atomici combinati.
Ibridizzazione sp3
Come evidenziato in (Figura 01.16-10), il mescolamento di un orbitale s e tre p produce quattro orbitali sp3 equivalenti, diretti verso i vertici di un tetraedro con angoli di circa 109,5°. In CH4, i quattro orbitali sp3 del carbonio (ognuno con un elettrone) si sovrappongono frontalmente con i 1s degli idrogeni generando quattro legami \(\sigma\) (Figura 01.16-11). La stessa ibridizzazione è coerente con la geometria di NF3 e H2O, dove la presenza di coppie solitarie su N e O deforma gli angoli rispetto all’ideale tetraedrico, mantenendo però la direzionalità sp3.
Ibridizzazione sp2
Secondo la (Figura 01.16-12), la combinazione di un s con due p porta a tre orbitali sp2 planari diretti a 120° (geometria trigonale planare), lasciando un p non ibridizzato perpendicolare al piano. Nell’etene, C2H4, i tre sp2 di ciascun C formano legami \(\sigma\) C–H e C–C nel piano (Figura 01.16-13), mentre i due orbitali p residui si sovrappongono lateralmente dando il legame \(\pi\) del doppio legame C=C (Figura 01.16-14). Un ulteriore esempio didatticamente equivalente è la formaldeide, H2C=O, in cui C è sp2: il doppio legame C=O consiste in un \(\sigma\) e in un \(\pi\) generato da p(C)–p(O).
Ibridizzazione sp
Come indicato in (Figura 01.16-15), l’ibridizzazione di un s con un p fornisce due orbitali sp lineari a 180°, con due p rimanenti non ibridizzati e mutuamente ortogonali. Nell’etino (acetilene), C2H2, gli sp di ciascun C formano legami \(\sigma\) C–H e C–C, mentre le due coppie di orbitali p si sovrappongono lateralmente generando due legami \(\pi\) tra i carboni (Figura 01.16-16). Anche CO2 illustra bene una configurazione sp sul carbonio, che spiega l’assetto lineare O=C=O e la presenza di due doppi legami equivalenti.
Altri schemi di ibridizzazione
Per geometrie non trattate dalle combinazioni sp, sp2 e sp3 si considerano schemi che coinvolgono orbitali d. In una descrizione VB classica:
- Quadrato planare: ibridizzazione dsp2 (talvolta indicata come sp2d), con quattro orbitali ibridi a 90° in un piano; tipica di complessi d8 a basso spin;
- Bipiramide trigonale: ibridizzazione sp3d, con cinque ibridi orientati verso i vertici di una bipiramide trigonale;
- Ottaedro: ibridizzazione d2sp3 o, in alternativa descrittiva, sp3d2, con sei ibridi diretti ai vertici di un ottaedro.
Questa formalizzazione è utile sul piano geometrico; in metalli di transizione pesanti, il coinvolgimento degli orbitali d di valenza superiori è spesso trattato in modo più accurato mediante teoria del campo dei leganti e del legame di gruppo, ma l’ibridizzazione resta un linguaggio operativo per correlare struttura e direzionalità dei legami.
Legame dativo e composti di coordinazione
Nei legami covalenti usuali ciascun atomo contribuisce con un elettrone. Esistono tuttavia legami in cui la coppia di legame proviene da un unico atomo donatore, mentre l’altro fornisce un orbitale vuoto a energia compatibile. Sono i legami dativi (o di coordinazione). Per esempio, lo ione ammonio, NH4+, si forma quando NH3 dona una coppia solitaria a H+, che offre un orbitale 1s vacante:
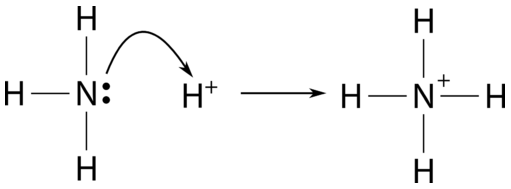 |
| Protonazione dell’ammoniaca – formazione dello ione ammonio (NH₄⁺) |
In modo analogo, H3O+ si origina dalla donazione di una coppia dall’ossigeno dell’acqua a H+:
| Protonazione dell’acqua – formazione dello ione idronio (H₃O⁺) |
La distinzione tra legame dativo e covalente “contributivo” è formale: le proprietà chimiche del legame risultante sono indistinguibili, ma la notazione aiuta a contabilizzare gli elettroni di valenza e le cariche formali. Secondo la terminologia di Lewis (vedi Capitolo 8), il donatore è una base di Lewis, l’accettore un acido di Lewis.
I legami di coordinazione sono centrali nei composti di coordinazione, costituiti da uno ione metallico centrale (elemento coordinante, spesso un metallo di transizione) e da ligandi dotati di coppie elettroniche disponibili. Lo ione complesso [Fe(CN)6]4− (esacianoferrato(II)) è un esempio classico: Fe2+ coordina sei anioni cianuro, ciascuno donatore di una coppia. Nel ferrocianuro di potassio, K4[Fe(CN)6], le quattro cariche negative dello ione complesso sono compensate da quattro K+. Tali sistemi non obbediscono alla regola dell’ottetto e coinvolgono direzionalità d-orbitali non trattate dai soli s e p.
Un ione complesso è caratterizzato da:
- uno ione centrale con orbitali di valenza vacanti, in grado di accogliere densità elettronica dai ligandi (acido di Lewis);
- ligandi dotati di coppie solitarie (basi di Lewis), con numero di coordinazione definito e geometria caratteristica;
- proprietà chimico-fisiche emergenti, differenti da quelle del metallo libero e dei ligandi isolati.
Le evidenze magnetiche e spettroscopiche di [Fe(CN)6]4− indicano un comportamento diamagnetico, quindi tutti gli elettroni risultano accoppiati. Per Fe2+ (Z = 26), con configurazione di valenza [Ar]3d6, la descrizione VB tradizionale postula che l’interazione con i sei CN− porti a un riassetto degli orbitali 3d, con formazione di sei orbitali ibridi d2sp3 orientati ottaedricamente (Figura 01.16-17) che accolgono le sei coppie donatrici. La geometria risultante è ottaedrica, con Fe2+ al centro e i sei ligandi ai vertici (Figura 01.16-18). L’accoppiamento elettronico è coerente con un complesso a basso spin, in accordo con la forte capacità accettrice di CN−.
Consideriamo ora [Fe(NH3)6]2+, esaamminoferro(II), che è paramagnetico. La descrizione VB lo interpreta con ibridizzazione sp3d2 (Figura 01.16-19), nella quale gli elettroni 3d rimangono in parte spaiati: il complesso è dunque ad alto spin. La differenza tra [Fe(CN)6]4− e [Fe(NH3)6]2+ riflette la diversa forza del campo dei ligandi (CN− più “forte” di NH3) e, conseguentemente, diverso grado di accoppiamento elettronico. Un’analogia di grande rilievo biologico è offerta dallo ione Fe2+ nell’eme dell’emoglobina: nella forma deossigenata si osserva un carattere paramagnetico (complesso esterno, alto spin), mentre nella forma ossigenata l’interazione con O2 promuove un assetto a più alto accoppiamento, assimilabile a un complesso interno con carattere a basso spin.
Le geometrie di coordinazione più comuni includono:
- ottaedrica, numero di coordinazione 6, descrivibile con d2sp3 o sp3d2;
- tetraedrica, numero di coordinazione 4, spesso razionalizzata tramite sp3;
- quadrato planare, numero di coordinazione 4, associata a dsp2 e tipica di complessi d8 a basso spin;
- bipiramide trigonale, numero di coordinazione 5, descrivibile da sp3d.
Osservazioni conclusive sulla robustezza del modello VB: la forza del legame è correlata alla sovrapposizione, rappresentabile dall’integrale di sovrapposizione \(S = \int \phi_A \phi_B \, d\tau\); una maggiore estensione radiale e una migliore collinearità degli orbitali incrementano \(S\) e, quindi, la stabilità del legame. Al contempo, la promozione e l’ibridizzazione hanno un costo energetico compensato dall’energia di legame guadagnata quando la geometria risultante massimizza la sovrapposizione \(\sigma\) e \(\pi\). La combinazione di considerazioni VB con la teoria del campo dei leganti offre una razionalizzazione coerente di geometrie, magnetismo e reattività dei complessi di metalli di transizione mantenendo la coerenza con i dati sperimentali citati in (Figura 01.16-08) e (Figura 01.16-19).
Image Gallery
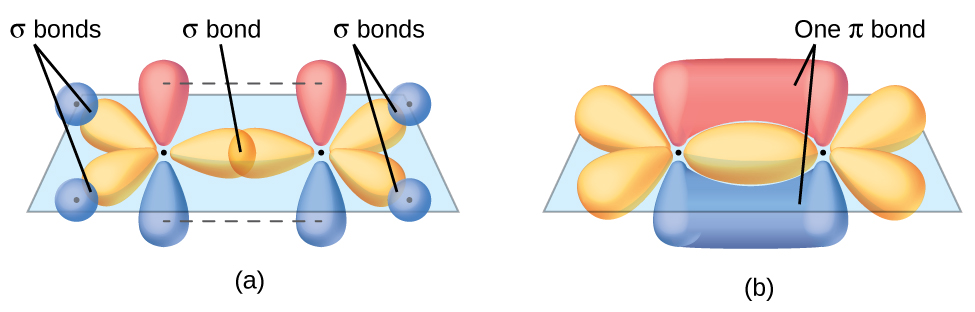
Image Gallery
Image Gallery

Image Gallery
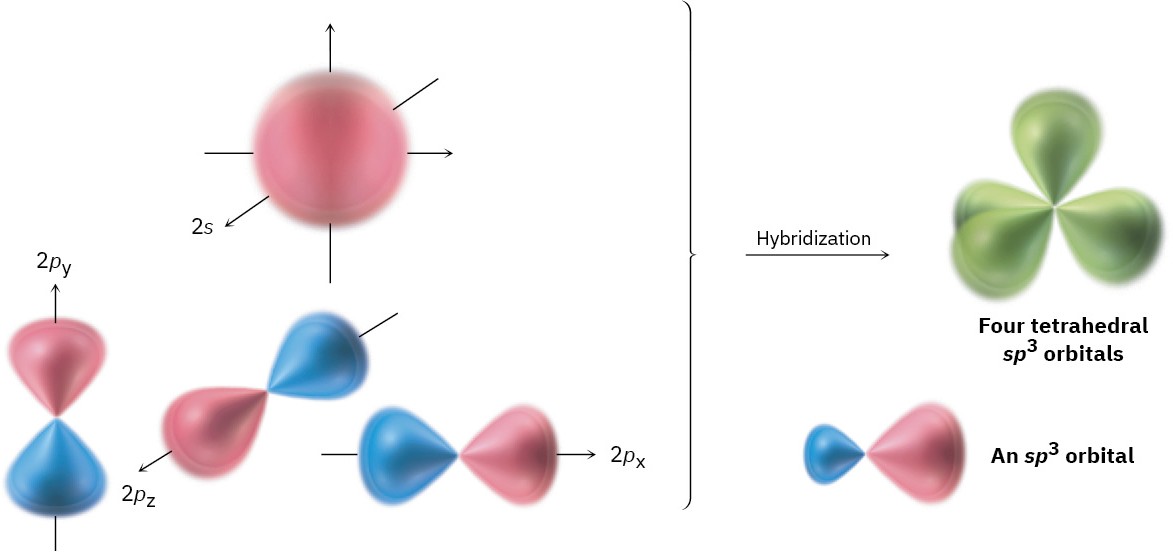
Image Gallery
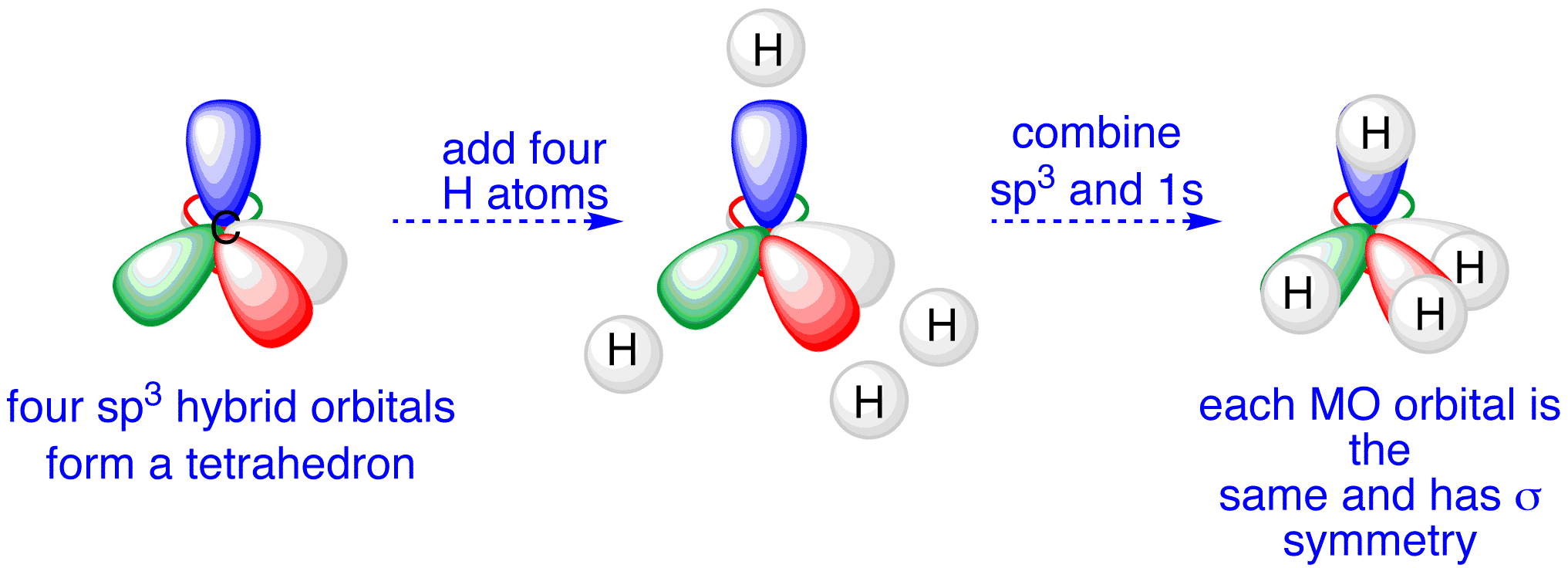
Image Gallery
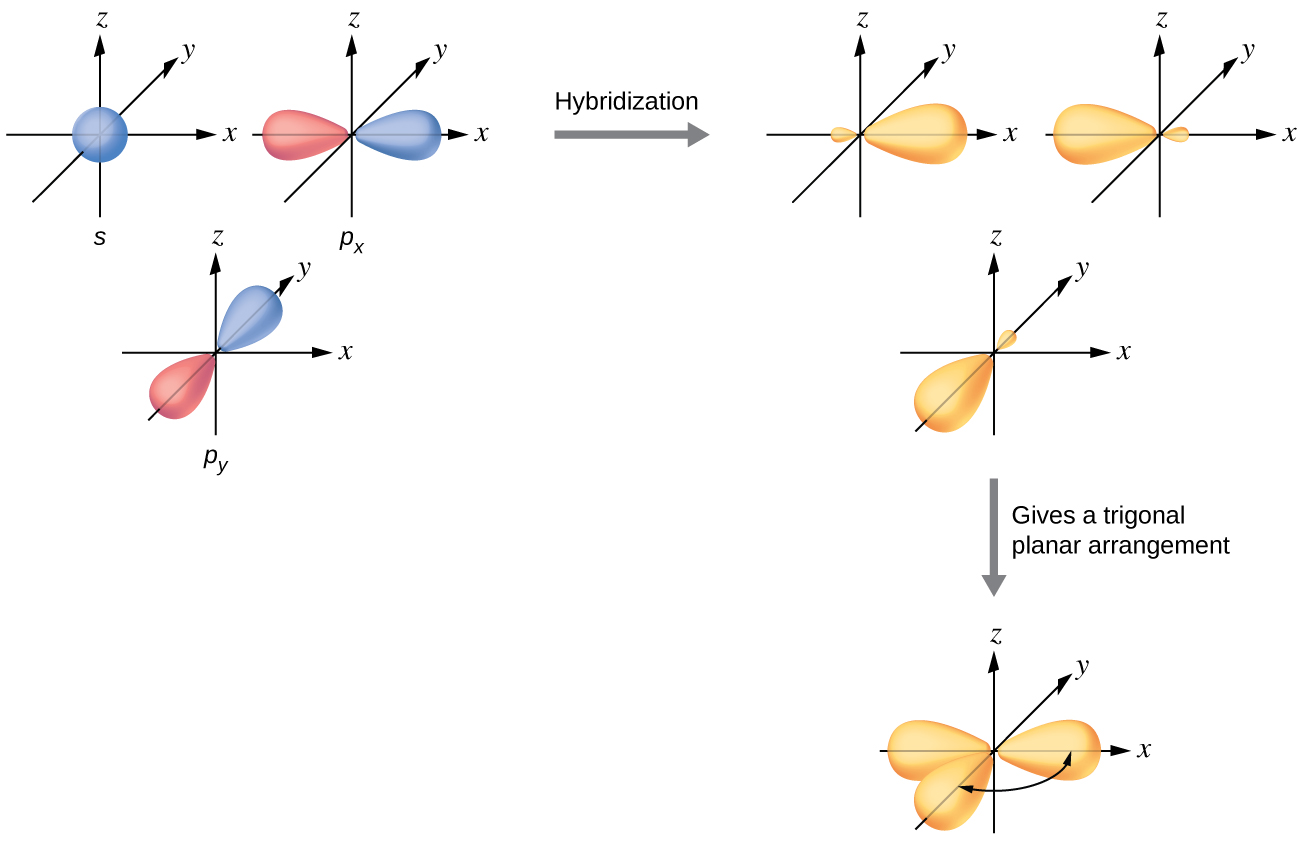
Image Gallery
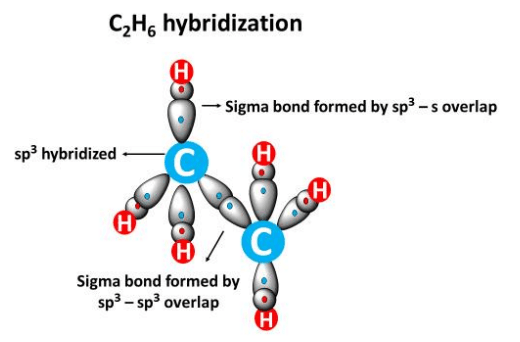
Image Gallery

Image Gallery

Image Gallery

Image Gallery

Image Gallery

Image Gallery

Image Gallery
Image Gallery


