


Risposta cellulare: la segnalazione cellulare conduce alla regolazione della trascrizione o delle attività citoplasmatiche
Definizione

L’informazione ereditaria è inscritta nell’ordine dei nucleotidi del DNA; per questo, la decifrazione della sequenza costituisce il presupposto per comprendere l’organizzazione dei geni, le loro modalità di regolazione e le relazioni evolutive tra specie. La lettura delle basi consente, inoltre, di associare varianti genomiche a fenotipi patologici umani, di progettare primer per la PCR e di ottenere, tramite clonaggio ed espressione, proteine in quantità. Il progresso tecnologico degli ultimi decenni ha trasformato il sequenziamento da procedura artigianale a processo ad alta produttività, riducendo drasticamente tempi e costi.
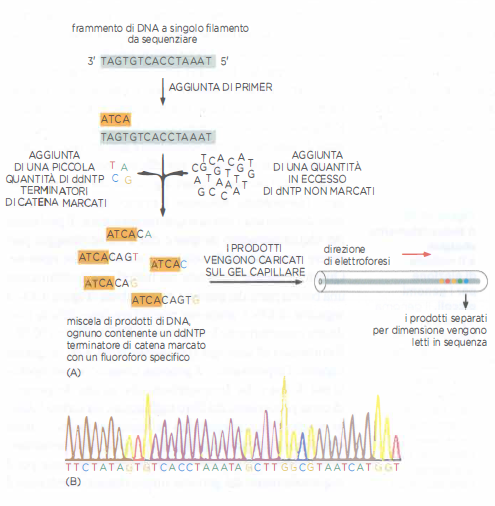
Alla fine degli anni ’70, il metodo a terminazione di catena (sequenziamento Sanger) ha reso possibile determinare con affidabilità la sequenza di frammenti di DNA purificati. Il principio si basa sull’impiego di dideossiribonucleosidi trifosfato (ddNTP), privi del gruppo 3’-OH: l’incorporazione di un ddNTP da parte della DNA polimerasi impedisce l’ulteriore estensione del filamento, generando prodotti che terminano in ogni posizione della sequenza bersaglio (Figura 04.09-01). A partire da un primer che ibrida sul DNA stampo, si ottiene così una popolazione di frammenti terminanti con A, C, G o T in tutti i punti possibili.
Il protocollo originario prevedeva reazioni separate e rivelazione su gel; l’implementazione moderna è completamente automatizzata. In capillari sottili, i prodotti vengono separati per dimensione e letti da un rivelatore che discrimina quattro fluorofori, ciascuno legato a un ddNTP di diversa base (Figura 04.09-02). Un software traduce la sequenza di picchi colorati nella serie di nucleotidi. L’accuratezza del metodo è elevata e le letture tipicamente raggiungono diverse centinaia di basi; per tali ragioni, il Sanger automatizzato ha avuto un ruolo centrale nel sequenziamento dei primi genomi umani e di numerosi organismi modello:
- Elementi chiave: DNA polimerasi ad alta fedeltà; ddNTP che bloccano l’estensione; primer specifico; separazione elettroforetica in capillare;
- Punti di forza: bassa frequenza di errore, utile per convalida di varianti e clonaggio;
- Limitazioni: produttività e costo per base sfavorevoli rispetto alle piattaforme più recenti.
Image Gallery
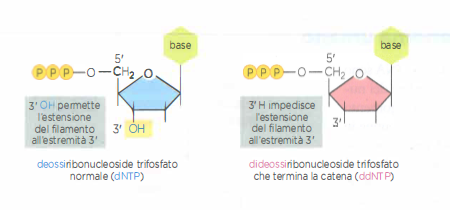
Image Gallery
Le tecnologie di sequenziamento di nuova generazione (NGS) hanno introdotto il concetto di lettura massivamente parallela, consentendo l’analisi simultanea di milioni di molecole. La preparazione delle librerie avviene senza passaggi clonali in batteri (Figura 04.09-04): il DNA genomico viene frammentato, le estremità sono riparate e sono aggiunti adattatori necessari per l’ancoraggio al supporto e per l’amplificazione. Ogni frammento è immobilizzato su un substrato solido (per esempio un vetrino funzionalizzato) o su una biglia; un’amplificazione in situ (per es. bridge amplification o PCR in emulsione) genera, in prossimità dello stampo, un insieme di copie identiche, formando un gruppo di molecole \( \approx 10^3 \) per sito. I gruppi vengono quindi letti in parallelo.
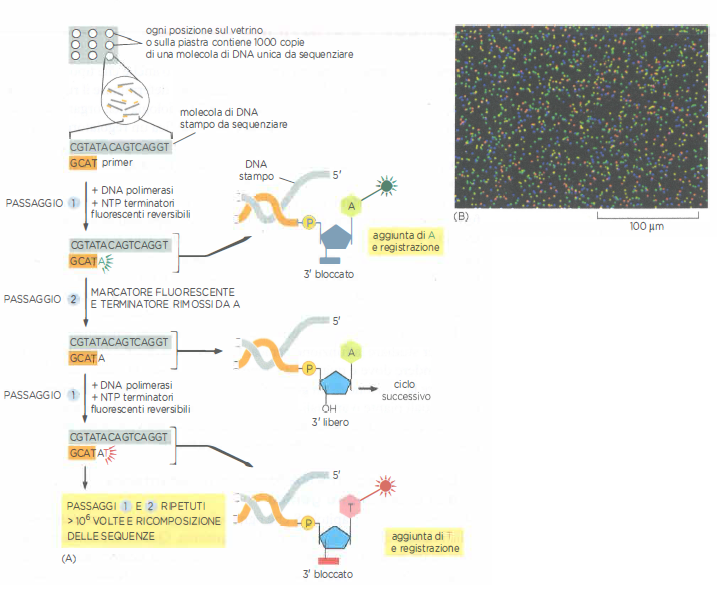
Una piattaforma ampiamente utilizzata è Illumina. Il ciclo di chimica si fonda su nucleotidi terminatori reversibili con etichette fluorescenti distinte: a ogni passo, la DNA polimerasi incorpora un singolo nucleotide modificato, si acquisisce un’immagine dell’intero vetrino per identificare, gruppo per gruppo, quale base è stata aggiunta, e poi si rimuovono il blocco e il fluoroforo per permettere il ciclo successivo (Figura 04.09-03). La molteplicità di reazioni in parallelo consente di sequenziare, in settimane, molti genomi con costi contenuti:
- Caratteristiche operative: letture in senso singolo o paired-end; indicizzazione (barcode) per multiplexing; controllo qualità tramite punteggi di base (es. Phred);
- Progettazione degli esperimenti: la copertura attesa si stima con la relazione di Lander–Waterman \( c = \frac{N \cdot L}{G} \), dove N è il numero di letture, L la loro lunghezza e G la dimensione del genoma; valori intorno a 30× sono comuni per il genoma umano;
- Gestione dei dati: allineamento al genoma di riferimento o assemblaggio de novo; chiamata di varianti; analisi di espressione e di interazioni DNA–proteina a seconda dell’applicazione;
- Fonti di bias: contenuto di GC estremo, regioni ripetute, errori specifici di piattaforma; mitigazione tramite design delle librerie, protocolli ottimizzati e filtraggio bioinformatico.
Le tecnologie di terza generazione abilitano la lettura di singole molecole senza amplificazione. Nel sequenziamento in tempo reale di singole molecole, una singola DNA polimerasi e il DNA stampo, appaiati a un primer, sono confinati in microcompartimenti ottici; l’incorporazione di dNTP marcati con fluorofori distinti viene registrata in tempo reale fino a ottenere l’intera sequenza dello stampo. Un’altra strategia guida la molecola di DNA attraverso un nanoporo di dimensioni sub-nanometriche: le quattro basi modulano in maniera caratteristica il segnale ionico durante il transito, permettendo l’assegnazione della sequenza. Questi approcci producono letture molto lunghe, utili per risolvere regioni ripetitive e riarrangiamenti strutturali, con correzione dell’errore ottenuta mediante coperture elevate o integrazione con dati NGS. Il continuo perfezionamento di chimiche, sensori e algoritmi sta ulteriormente riducendo tempi e costi del sequenziamento dell’intero genoma umano.
Image Gallery
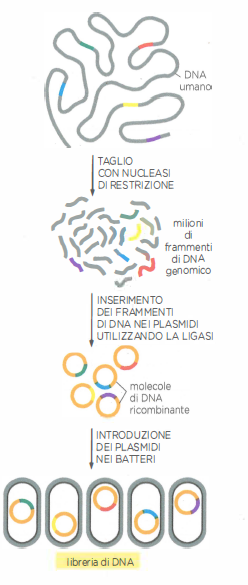
Image Gallery
Una sequenza grezza di basi, di per sé, non descrive come l’informazione genetica si traduca in funzioni cellulari o in tratti organismici. Le banche dati pubbliche e gli algoritmi di allineamento permettono di confrontare la sequenza in esame con repertori annotati, così da inferire la presenza di geni e prevederne il ruolo sulla base degli omologhi noti in altri taxa. Le regioni codificanti mostrano, in generale, maggiore conservazione rispetto a vaste porzioni non codificanti, sebbene esistano elementi regolativi e RNA non codificanti che risultano conservati per vincoli funzionali:
- Individuazione dei geni: ricerca di open reading frame, segnali di splicing, codon usage e motivi promotori;
- Annotazione funzionale: allineamenti a proteine con domini noti, identificazione di ortologhi e paraloghi, analisi di synteny tra genomi correlati;
- Evoluzione e filogenesi: stima delle distanze tra sequenze, costruzione di alberi filogenetici e datazione di divergenze su set di geni conservati;
- Identificazione di campioni ignoti: assegnazione tassonomica di frammenti provenienti da suolo, acqua marina o campioni clinici (metagenomica), utile per riconoscere agenti infettivi non diagnosticati e per ricostruire comunità microbiche;
- Prioritizzazione di varianti: confronto con cataloghi di polimorfismi e mutazioni patogeniche, valutazione della conservazione interspecifica per inferire l’impatto funzionale.
Le analogie di sequenza aiutano a ricondurre un frammento alla sua origine e a identificare specie strettamente imparentate, mentre le differenze forniscono indizi sui meccanismi evolutivi e sui tratti soggetti a selezione. La combinazione di dati di sequenza, strutture geniche e contesto genomico rappresenta la base dell’annotazione affidabile e dell’interpretazione biologica delle varianti.


