Proprietà delle soluzioni che dipendono dalla concentrazione: proprietà colligative
Definizione

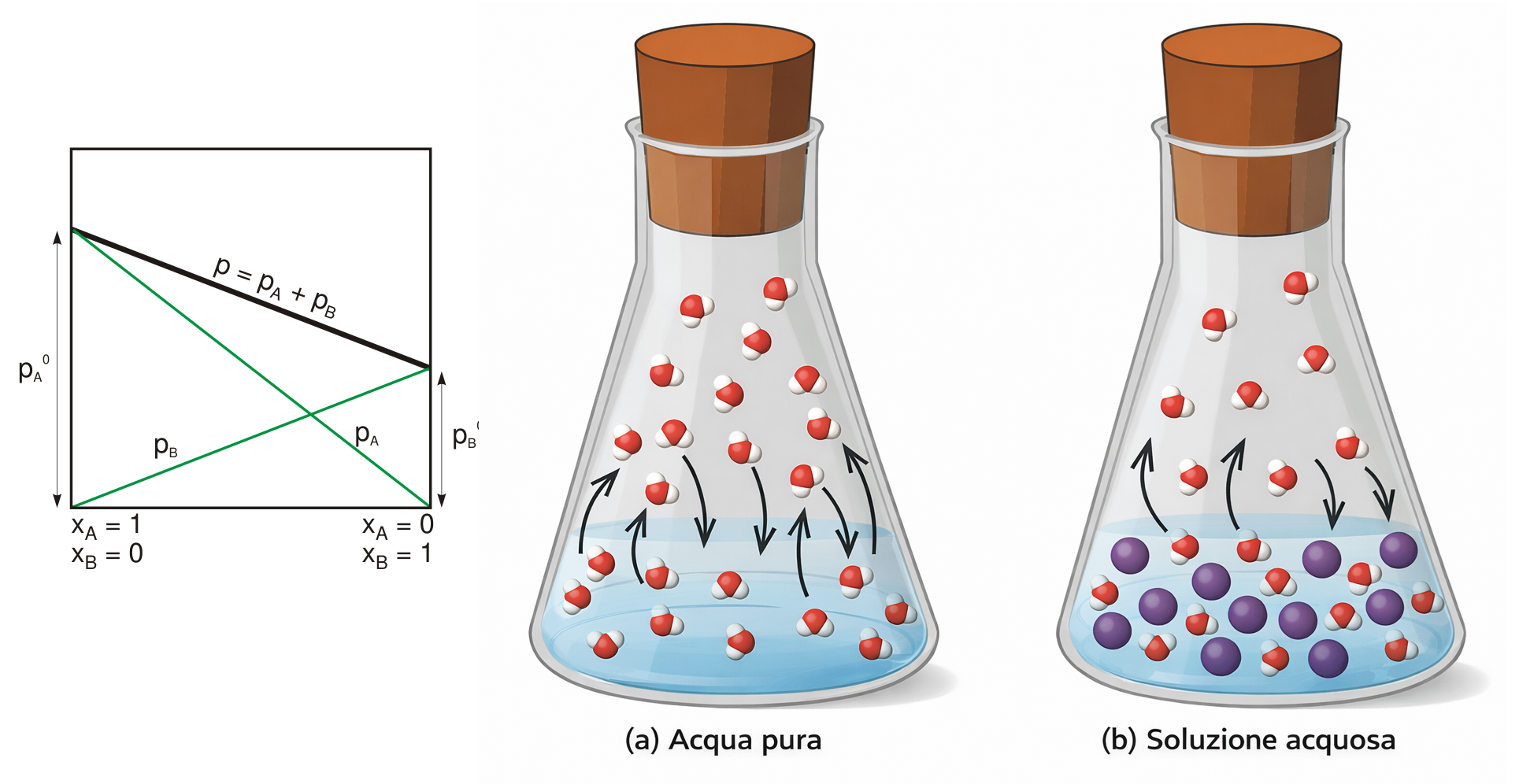
La legge di Raoult per soluzioni ideali enuncia che, in presenza di un soluto non volatile, la pressione di vapore del solvente diminuisce in proporzione alla sua frazione molare. Se il solvente puro ha pressione di vapore \(p_A^{\ast}\) e la sua frazione molare in soluzione è \(x_A\), allora \(p_A = x_A\, p_A^{\ast}\); per un soluto non volatile vale dunque \(\Delta p = p_A^{\ast} - p_A = x_B\, p_A^{\ast}\), con \(x_B\) frazione molare del soluto. La conseguenza macroscopica più rilevante di questa relazione è la modifica dei punti di transizione di fase: la temperatura di solidificazione si abbassa, mentre la temperatura di ebollizione si innalza.
Una lettura molecolare del fenomeno chiarisce l’origine della diminuzione di pressione di vapore. L’evaporazione richiede che molecole di solvente lascino la superficie del liquido, raggiungendo lo stato di vapore sino all’equilibrio dinamico alla data temperatura. Le particelle di soluto, occupando siti alla superficie e alterando la distribuzione delle energie, riducono il numero di molecole di solvente che riescono a sfuggire per unità di tempo; la pressione di vapore di equilibrio risulta quindi inferiore rispetto al solvente puro (Figura 03.04-01). Per contro, all’aumentare della temperatura cresce la pressione di vapore della soluzione finché uguaglia la pressione esterna: ciò richiede una temperatura maggiore rispetto al solvente puro, come discusso più avanti.
Image Gallery
L’equilibrio tra fase liquida e fase solida dell’acqua al punto di congelamento può essere rappresentato da \(\mathrm{H_2O(l)\rightleftharpoons H_2O(s)}\). La presenza di un soluto non volatile abbassa il potenziale chimico del solvente nella fase liquida più di quanto non lo faccia nella fase solida (nella quale il soluto è, in prima approssimazione, escluso dal reticolo). Per ristabilire l’uguaglianza dei potenziali chimici, l’equilibrio si sposta a temperatura più bassa, da cui la depressione crioscopica. Un ragionamento simmetrico vale per l’ebollizione: poiché il soluto riduce la pressione di vapore del liquido, è necessaria una temperatura più alta affinché \(p_{\text{vapore}}\) eguagli la pressione atmosferica, e il punto di ebollizione aumenta.
In regime diluito, le variazioni dei punti di transizione sono proporzionali alla concentrazione molale del soluto. Per un soluto non dissociante si hanno: \[ \Delta T_c = K_c\, m,\qquad \Delta T_{eb} = K_{eb}\, m, \] dove \(m\) è la molalità, \(K_c\) la costante crioscopica e \(K_{eb}\) la costante ebullioscopica del solvente. Per tenere conto della dissociazione o dell’associazione molecolare dei soluti, si introduce il fattore di van ’t Hoff \(i\) (numero effettivo di particelle per unità formula in soluzione), ottenendo le forme generali: \[ \Delta T_c = K_c\, m\, i,\qquad \Delta T_{eb} = K_{eb}\, m\, i. \] Per non elettroliti \(i \approx 1\); per elettroliti forti in soluzioni molto diluite \(i\) si avvicina al numero di ioni generati per unità formula, con lievi deviazioni dovute all’interazione ionica.
La natura colligativa di questi effetti dipende dal numero di particelle, non dalla loro massa: a parità di moli di particelle totali in soluzione, molecole “pesanti” e “leggere” producono lo stesso effetto su \(\Delta T_c\) e \(\Delta T_{eb}\). Per questo motivo si usano unità legate alle moli e non alla massa. La molalità, definita come moli di soluto per chilogrammo di solvente, è preferita perché non varia con la temperatura, a differenza della molarità.
La distinzione tra elettroliti e non elettroliti è cruciale. Un non elettrolita come il glucosio si dissolve senza dissociarsi: \[ \ce{C6H12O6(s) ->[H2O] C6H12O6(aq)}, \] così 1 mol in 1 kg di acqua fornisce \(1\) mol di particelle. Un elettrolita forte come il cloruro di sodio si dissocia in ioni: \[ \ce{NaCl(s) ->[H2O] Na+(aq) + Cl-(aq)}, \] e idealmente 1 mol genera circa 2 mol di particelle; un sale come \(\ce{CaCl2}\) può liberare circa 3 mol di ioni per mol. In soluzioni reali, soprattutto a concentrazioni non infinitesime, l’effettivo \(i\) può essere leggermente inferiore a causa dell’appaiamento ionico e di interazioni a corto raggio:
- bagnimaria a salamoia (ad esempio con \(\ce{NaCl}\) o \(\ce{CaCl2}\)) impiegati nella produzione artigianale di gelati per ottenere temperature inferiori a 0 °C senza congelare completamente il bagno termostatico;
- fluidi lavavetri a base di etanolo o isopropanolo, formulati per ridurre il punto di congelamento e innalzare quello di ebollizione, garantendo funzionalità in un ampio intervallo di temperature;
- miscele acqua–glicole propilenico nei circuiti HVAC industriali, che ampliano la finestra operativa evitando sia il gelo invernale sia ebollizioni locali nelle zone a più alto carico termico.
Le costanti crioscopica ed ebullioscopica dipendono esclusivamente dal solvente. Per l’acqua valgono: \[ K_c = 1,86 \ \frac{^\circ\mathrm{C}\cdot \mathrm{kg}}{\mathrm{mol}} \ ;\quad K_{eb} = 0,512 \ \frac{^\circ\mathrm{C}\cdot \mathrm{kg}}{\mathrm{mol}}. \] Conoscendo la molalità \(m\) del soluto e il fattore di van ’t Hoff \(i\), si calcolano le variazioni di temperatura mediante \(\Delta T_c = K_c m i\) e \(\Delta T_{eb} = K_{eb} m i\). Ricordando che, a 1 atm, l’acqua pura congela a 0 °C e bolle a 100 °C, i punti della soluzione si ottengono come: \[ T_{\text{cong, sol}} = 0\,^\circ\mathrm{C} - \Delta T_c,\qquad T_{\text{eb, sol}} = 100\,^\circ\mathrm{C} + \Delta T_{eb}. \]
Esempio numerico. Si confrontino due soluzioni acquose 0,200 m a 1 atm: una di urea (\(\ce{NH2CONH2}\), non elettrolita, \(i \approx 1\)) e una di \(\ce{NaCl}\) con \(i \approx 2{,}0\) in regime diluito:
- Urea: \(\Delta T_c = 1{,}86 \times 0{,}200 \times 1 = 0{,}372\ ^\circ\mathrm{C}\); \(\Delta T_{eb} = 0{,}512 \times 0{,}200 \times 1 = 0{,}102\ ^\circ\mathrm{C}\). Pertanto \(T_{\text{cong}} \approx -0{,}372\ ^\circ\mathrm{C}\) e \(T_{\text{eb}} \approx 100{,}102\ ^\circ\mathrm{C}\);
- \(\ce{NaCl}\): \(\Delta T_c = 1{,}86 \times 0{,}200 \times 2{,}0 = 0{,}744\ ^\circ\mathrm{C}\); \(\Delta T_{eb} = 0{,}512 \times 0{,}200 \times 2{,}0 = 0{,}205\ ^\circ\mathrm{C}\). Quindi \(T_{\text{cong}} \approx -0{,}744\ ^\circ\mathrm{C}\) e \(T_{\text{eb}} \approx 100{,}205\ ^\circ\mathrm{C}\).
I valori sperimentali per elettroliti possono risultare leggermente diversi da quelli ideali: un \(i\) effettivo minore di 2,0 riflette interazioni ioniche e deviazioni dall’idealità. In generale, l’accuratezza della legge di Raoult e delle espressioni crioscopiche/ebullioscopiche è massima per soluzioni diluite, soluti non volatili e sistemi prossimi all’idealità; per concentrazioni maggiori occorre introdurre attività e coefficienti di attività per una descrizione quantitativa più rigorosa.
La membrana plasmatica regola in modo selettivo lo scambio di materia tra citoplasma e ambiente, consentendo il passaggio controllato di determinate specie chimiche e opponendosi ad altre. Un meccanismo fondamentale di trasporto è la diffusione, ossia il moto netto di molecole dal compartimento a concentrazione più elevata verso quello a concentrazione inferiore. Il gradiente di concentrazione è la differenza di concentrazione di una medesima sostanza tra due volumi adiacenti separati, ad esempio, da una membrana. A causa della sua struttura lipidica e delle proteine associate, la membrana cellulare è selettivamente permeabile: piccole molecole apolari e, in parte, molecole polari molto piccole possono attraversarla più facilmente, mentre macromolecole e ioni sono generalmente esclusi in assenza di canali o trasportatori dedicati.
Quando i soluti non possono oltrepassare la barriera, è il solvente a muoversi. Una membrana che consente il passaggio del solvente ma non del soluto si definisce semipermeabile. L’osmosi è il flusso netto del solvente attraverso una membrana semipermeabile in risposta a un gradiente del potenziale chimico dell’acqua, che nei sistemi biologici equivale in larga parte a un gradiente di concentrazione dell’acqua stessa.
Si consideri una sacca da dialisi contenente una soluzione di glucosio 0,5 M, immersa in un becher con acqua pura. I pori della membrana lasciano passare l’acqua, ma non le molecole di glucosio. La concentrazione di glucosio è maggiore all’interno, ma il glucosio non può diffondere per annullare il gradiente; viceversa, la concentrazione di acqua è più alta all’esterno che all’interno, e pertanto l’acqua diffonde nella sacca secondo il suo gradiente. Il processo, rappresentato schematicamente in (Figura 03.04-02), prosegue finché la distensione della membrana genera una controforza sufficiente a bilanciare l’ingresso netto di acqua.
La pressione osmotica è la pressione esterna che occorrerebbe applicare alla soluzione per arrestare il flusso osmotico del solvente attraverso la membrana semipermeabile. In termini operativi, è la pressione con cui l’acqua passerebbe per osmosi dal compartimento di acqua pura al compartimento contenente la soluzione, in presenza della membrana semipermeabile.
Per soluzioni diluite la pressione osmotica obbedisce alla legge di van ’t Hoff, formalmente analoga alla legge dei gas ideali. Partendo dalla relazione dei gas ideali \( PV = nRT \), si ottiene l’equazione osmotica sostituendo la pressione P con la pressione osmotica \( \pi \):
\[ \pi V = nRT \quad \Longrightarrow \quad \pi = \frac{n}{V}RT = CRT, \]
dove \( C = n/V \) è la molarità della soluzione, R la costante universale dei gas e T la temperatura assoluta. Per tener conto della dissociazione degli elettroliti si introduce il fattore di van ’t Hoff \( i \) (numero effettivo di particelle in soluzione per unità di formula alla diluizione considerata):
\[ \pi = iMRT. \]
La pressione osmotica è una proprietà colligativa: dipende dal numero totale di particelle di soluto in soluzione e non dalla loro natura chimica. Per un non elettrolita come il glucosio, \( i \approx 1 \); per un elettrolita binario ideale come NaCl, \( i \approx 2 \); per CaCl\(_2\), \( i \approx 3 \). In soluzioni non estremamente diluite, associazioni ioniche e interazioni specifiche possono ridurre \( i \) rispetto al valore intero teorico.
È utile collegare questa relazione al concetto di osmolarità, definita come la molarità delle particelle osmoticamente attive: per un singolo soluto, \( \text{osmolarità} = iM \). In miscele di soluti, l’osmolarità totale è la somma delle osmolarità parziali. Per completezza, si ricorda che l’osmolalità esprime analogo contenuto per chilogrammo di solvente e risulta meno influenzata dalle variazioni di volume con la temperatura.
Dato che \( \pi \) è proporzionale a T, la temperatura influisce direttamente sulla pressione osmotica. Ad esempio, per una soluzione 0,20 M di saccarosio a 298 K, con \( i = 1 \) e \( R = 0,08206 \, \text{L·atm·mol}^{-1}\text{·K}^{-1} \), si ha \( \pi \approx 0,20 \times 0,08206 \times 298 \approx 4,88 \, \text{atm} \).
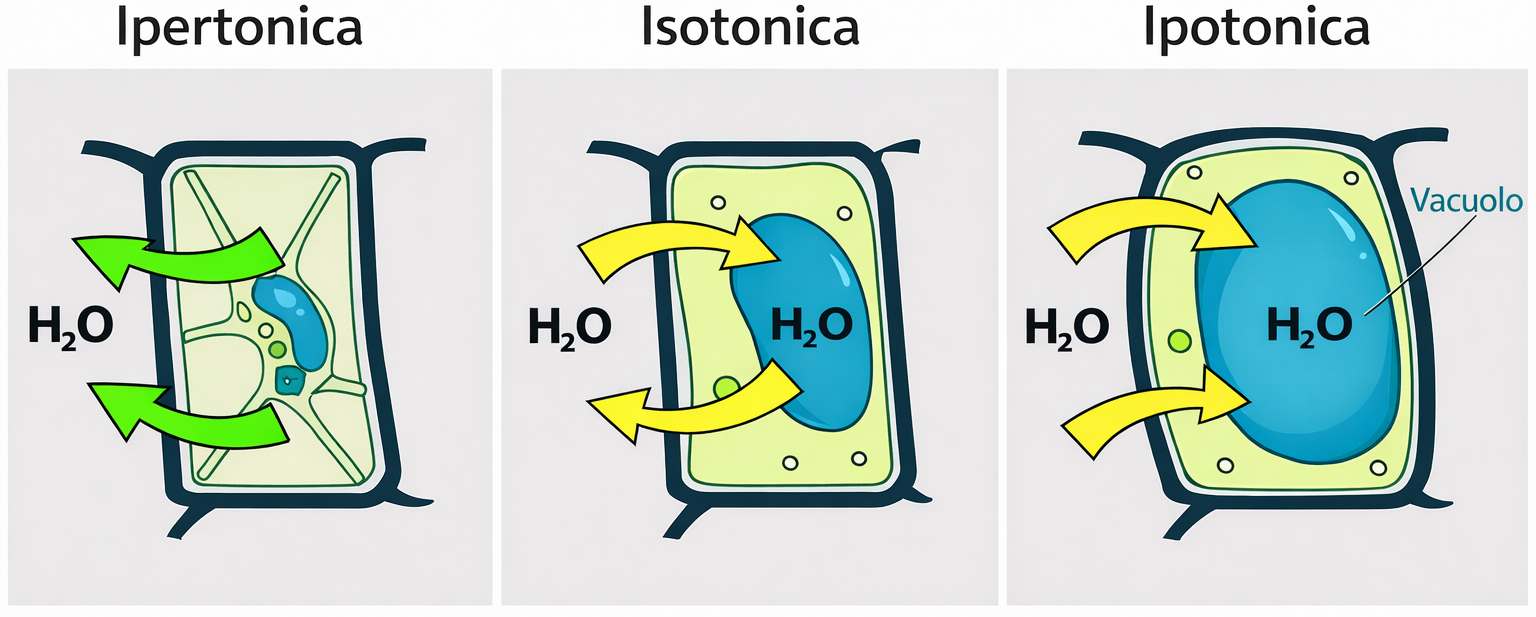
Nel sangue, l’osmolarità del plasma è approssimativamente equivalente a quella di una soluzione 0,30 M di glucosio oppure a quella di una soluzione 0,15 M di NaCl, poiché il cloruro di sodio si dissocia fornendo circa il doppio di particelle rispetto a un non elettrolita a parità di molarità. I globuli rossi, aventi osmolarità simile al plasma, non mostrano variazioni di volume in una soluzione isotonica, cioè con uguale pressione osmotica efficace: in tal caso la cellula mantiene forma e dimensioni (Figura 03.04-03).
Se il mezzo esterno è ipotonico rispetto al citoplasma (osmolarità più bassa), l’acqua entra nella cellula e, data la ridotta resistenza meccanica della membrana eritrocitaria, può verificarsi rigonfiamento fino alla lisi osmotica o emolisi (Figura 03.04-03). Viceversa, in un ambiente ipertonico (osmolarità più alta), l’acqua esce dalla cellula con conseguente contrazione del volume e aspetto crenato (Figura 03.04-03). In ambito fisiologico, la tonicitá dipende dai soluti non penetranti: soluti che attraversano rapidamente la membrana (ad esempio l’urea) hanno minore effetto sulla pressione osmotica efficace. Più in generale, l’efficacia osmotica attraverso una membrana biologica parzialmente permeabile può essere descritta con il coefficiente di riflessione \( \sigma \) (0 ≤ \( \sigma \) ≤ 1), per cui la spinta osmotica effettiva vale \( \pi_{\text{eff}} = \sigma \, iMRT \).
Questi concetti hanno ricadute cliniche immediate nella scelta dei fluidi infusionali (Figura 03.04-04). Un liquido endovenoso deve risultare isotonico rispetto al plasma e agli eritrociti per non alterare il bilancio idrico tra compartimento intravascolare e cellule. Sono comunemente impiegate soluzioni di destrosio (glucosio) al 5,5% in massa/volume, pari a circa 0,30 M, e soluzioni saline “normali” allo 0,9% di NaCl, corrispondenti a circa 0,15 M: entrambe presentano, a temperatura fisiologica, una pressione osmotica prossima a quella del plasma e possono essere somministrate senza indurre variazioni significative del volume eritrocitario.
La differenza tra osmolarità e tonicitá è didatticamente cruciale: due soluzioni con uguale osmolarità possono non essere isotoniche se parte dei soluti attraversa la membrana. Per questo motivo, nelle soluzioni infusionali si privilegiano soluti non penetranti (Na\(^+\), Cl\(^-\)) o che diventano rapidamente metabolizzati in modo controllato (destrosio), così da non generare shock osmotici a livello cellulare.
Alcune applicazioni ed esempi quotidiani di osmosi includono i casi seguenti:
- Ingestione di acqua marina: l’elevata concentrazione di sali rende il fluido ipertonico rispetto ai liquidi corporei, favorendo la fuoriuscita di acqua dalle cellule e aggravando la disidratazione;
- Fette di cetriolo cosparse di sale che perdono turgore: l’ambiente ipertonico esterno richiama acqua dai tessuti vegetali, con raggrinzimento visibile (Figura 03.04-05);
- Osmosi inversa nei sistemi di dissalazione: applicando una pressione superiore alla pressione osmotica si forza il passaggio dell’acqua pulita attraverso una membrana, lasciando i sali nel lato ad alta concentrazione;
- Terapia di reidratazione orale: soluzioni contenenti sodio e glucosio sfruttano il cotrasporto Na\(^+\)/glucosio per incrementare l’assorbimento d’acqua nell’epitelio intestinale, ripristinando l’equilibrio osmotico in condizioni di diarrea acuta.
Per completezza, si riassumono alcune equivalenze utili al calcolo: una soluzione 1,0 M di glucosio fornisce 1,0 osmol/L; una soluzione 1,0 M di NaCl fornisce idealmente 2,0 osmol/L; una soluzione 1,0 M di CaCl\(_2\) apporta idealmente 3,0 osmol/L. In pratica, a concentrazioni non infinitesimali si osservano scostamenti dovuti a interazioni ioniche, ma per scopi fisiologici e didattici le approssimazioni ideali risultano spesso adeguate.
Image Gallery
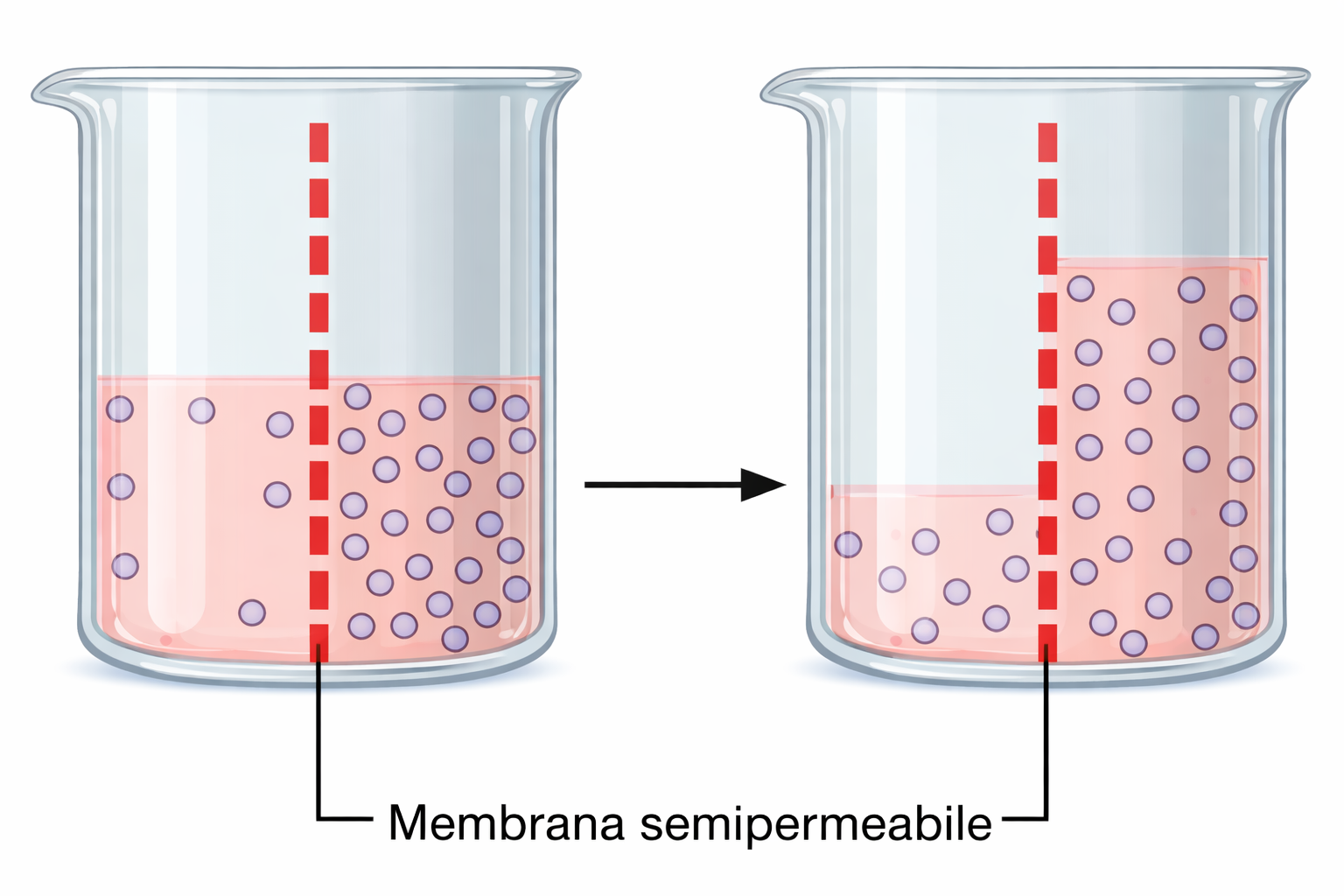
Image Gallery
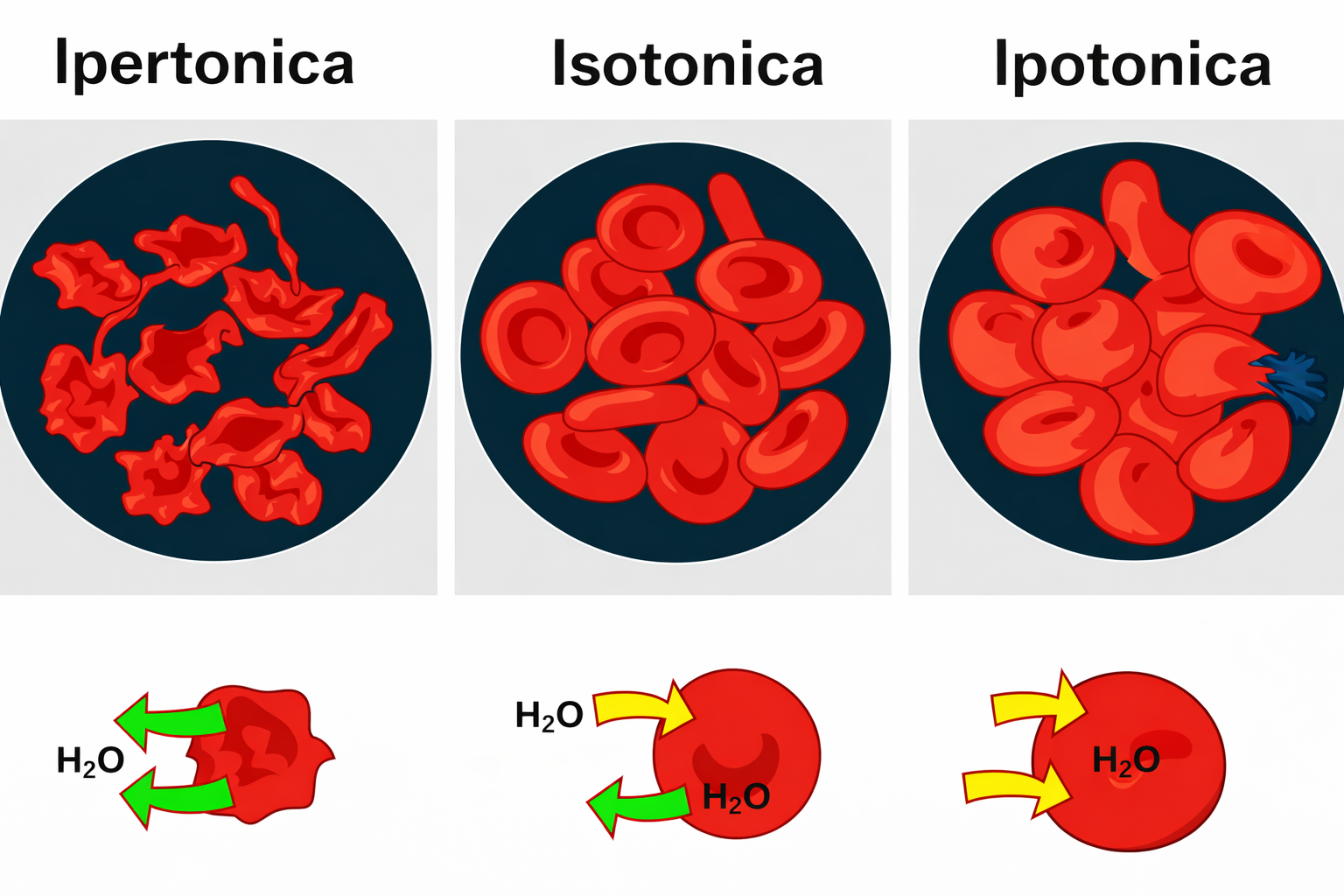
Image Gallery
Image Gallery
Image Gallery

Image Gallery