L’apoptosi richiede l’interazione di molteplici vie di segnalazione cellulare
TOPICS
Definizione

Identificare l’origine di una sequenza di acidi nucleici e inferirne la potenziale funzione costituisce un punto di partenza, non il traguardo, per comprenderne il contributo allo sviluppo, all’omeostasi e alla fisiopatologia dell’organismo. Sapere che un tratto di DNA codifica un fattore di trascrizione, ad esempio, non chiarisce in quali cellule ne venga prodotta la proteina, in quali fasi temporali essa compaia né quali bersagli regolativi controlli. Per rispondere a queste domande occorre un’indagine sperimentale, fondata su scelte metodologiche spesso modellate da competenze ed esperienze del ricercatore o della ricercatrice: una genetista può ricorrere a approcci di perdita o guadagno di funzione in organismi modello, mentre un biochimico può produrre elevate quantità della proteina per determinarne struttura e proprietà fisico-chimiche. Gli approcci attuali alla funzione genica combinano strumenti complementari: misure dell’espressione a livello di RNA, mappatura spaziale delle trascrizioni, tracciamento in vivo delle proteine e manipolazioni mirate del genoma o del trascrittoma. Tecniche come l’editing con CRISPR-Cas, l’inattivazione condizionale, l’overespressione controllata, insieme all’espressione eterologa e alla purificazione proteica, permettono di collegare sequenza, espressione, localizzazione e fenotipo.
Ogni tipologia cellulare manifesta solo una frazione del repertorio genico totale, e questa scelta di espressione varia in funzione del tipo cellulare, dello stato fisiologico e del microambiente. La determinazione dei trascritti presenti in una popolazione cellulare o in un tessuto fornisce una stima del trascrittoma, ossia l’insieme degli RNA prodotti in condizioni definite. Le tecnologie di sequenziamento di nuova generazione consentono oggi di misurare in modo estensivo e quantitativo tali profili, con risoluzione fino al singolo trascritto.
L’approccio comunemente usato, denominato RNA‑Seq, prevede che gli RNA estratti vengano convertiti in cDNA mediante trascrittasi inversa, quindi preparati come librerie di sequenziamento. Strategie di arricchimento come la selezione per code poli(A) o la deplezione degli rRNA riducono il rumore di fondo, mentre l’uso di identificatori molecolari univoci (UMI) migliora la quantificazione riducendo i bias di amplificazione. Il sequenziamento, seguito dall’allineamento a un genoma o trascrittoma di riferimento, permette di stimare l’abbondanza dei trascritti con misure normalizzate quali TPM o CPM.
Oltre a quantificare i livelli di espressione, l’RNA‑Seq rileva eventi di splicing alternativo, isoforme di inizio e fine trascrizionale, trascritti a bassa abbondanza, varianti alleliche espresse e RNA non codificanti. Approcci a lettura lunga possono risolvere isoforme complesse, mentre disegni sperimentali con replicati biologici e modelli statistici adeguati (ad esempio, basati su distribuzioni binomiali negative) consentono di identificare differenze significative tra condizioni, trattamenti farmacologici e genotipi.
Una limitazione intrinseca delle misure su popolazioni cellulari è la perdita dell’eterogeneità. Per questo, il sequenziamento di singole cellule ha acquisito rilievo: permette di distinguere sottopopolazioni, stati transitori e traiettorie differenziative, mantenendo però solo una mappa inferenziale dello spazio, non una localizzazione fisica. In tale quadro, le informazioni spaziali sono fornite da metodiche complementari descritte di seguito:
- domande affrontabili con l’RNA‑Seq: quali geni cambiano espressione tra due condizioni; quali isoforme dominano in un dato tessuto; quali vie biologiche risultano arricchite; quali trascritti rari emergono in risposta a uno stimolo;
- accortezze sperimentali: controllo dei batch, adeguata profondità di lettura, qualità dell’RNA, scelte di normalizzazione, uso di controlli esterni (spike‑in) quando opportuno.
Questa tecnologia ha trasformato lo studio delle dinamiche dell’espressione lungo lo sviluppo, durante il ciclo cellulare, in reazione a perturbazioni chimiche o genetiche e in diversi distretti tissutali, fornendo un quadro quantitativo dell’attività genica in contesti fisiologici e patologici.
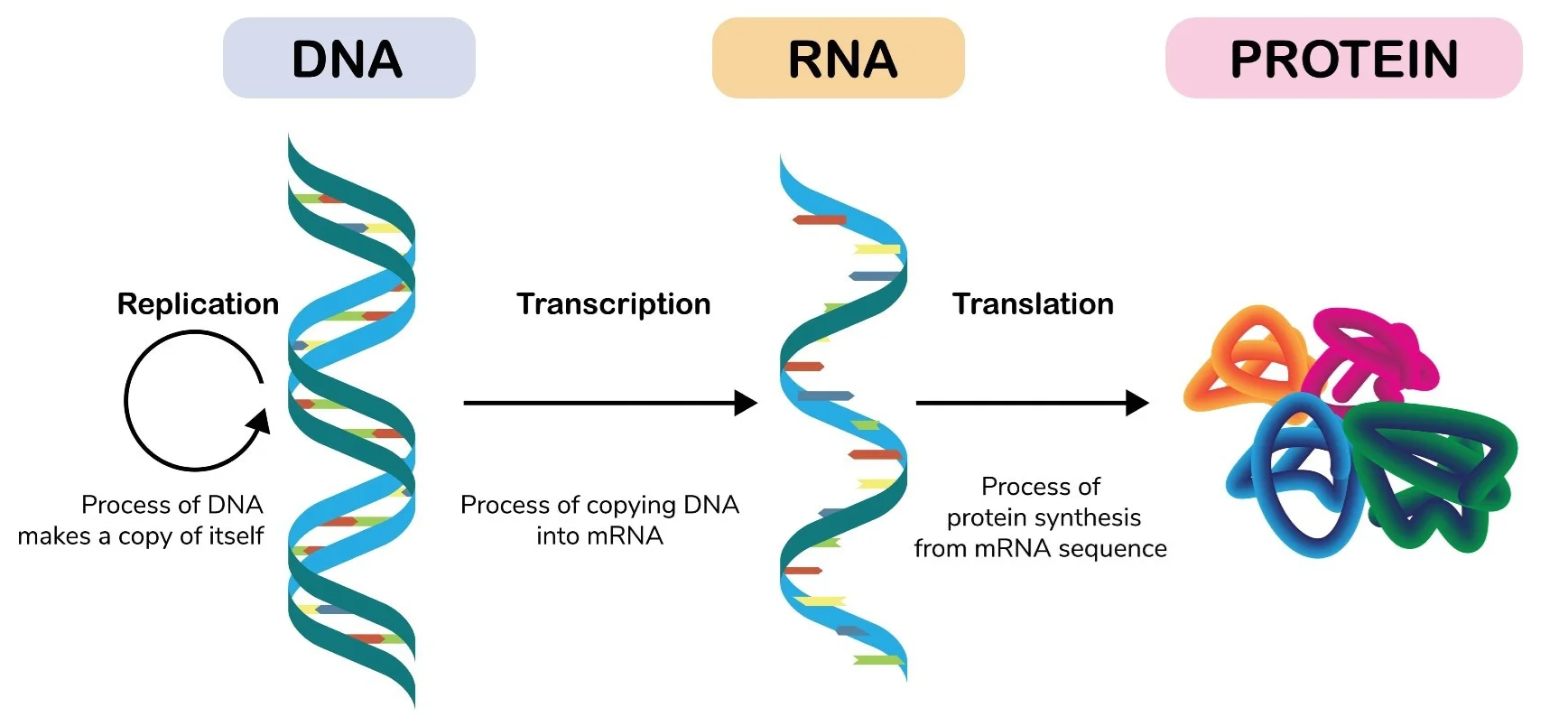
Analisi globali come microarray ed RNA‑Seq definiscono quali geni siano attivi in un campione, ma non indicano in quale compartimento cellulare o in quali cellule specifiche i trascritti vengano prodotti. L’ibridazione in situ colma questa lacuna, visualizzando direttamente, nella sede nativa, sequenze specifiche di DNA o RNA mediante sonde complementari marcate. In pratica, tessuti o cellule vengono fissati e permeabilizzati, quindi esposti a sonde a singolo filamento (DNA o RNA) etichettate con fluorofori o isotopi. Dopo lavaggi selettivi, il segnale rivelato indica la posizione dei trascritti target. A livello cellulare e tissutale, l’ibridazione in situ consente di mappare domini di espressione e gradienti (Figura 04.10-01); a livello cromosomico, l’ibridazione in situ fluorescente (FISH) su cromosomi metafasici o nuclei interfasici permette di individuare riarrangiamenti, delezioni o anomalie numeriche, impiegate ad esempio nella diagnosi prenatale di aneuploidie (Figura 04.10-02). Varianti ad alta sensibilità, come la single‑molecule FISH (smFISH) o approcci chimici ottimizzati, consentono il conteggio quasi molecola‑per‑molecola dei trascritti e la multiplexazione di molteplici target nello stesso campione. Tali metodiche, combinate con microscopia a elevata risoluzione, rivelano pattern spazio‑temporali di espressione, interazioni tra cellule vicine e precise transizioni di stato:
- vantaggi: localizzazione subcellulare dei trascritti, possibilità di correlare espressione e morfologia, compatibilità con marcatori proteici;
- controlli essenziali: sonde senso/antisenso, trattamento con RNasi come controllo negativo, sonde contro trascritti housekeeping per la qualità dell’RNA;
- limiti e mitigazioni: necessità di campioni ben fissati, rischio di segnali aspecifici (riducibile ottimizzando condizioni di ibridazione), capacità di multiplex limitata ma in crescita con strategie codificate.
La scelta tra un’analisi trascrittomica bulk o single‑cell e l’ibridazione in situ dipende dal quesito: quantificazione globale e confronti statistici trovano nell’RNA‑Seq lo strumento principale; la definizione della “geografia” dell’espressione richiede l’ibridazione in situ, spesso integrata con immagini di riferimento e marcatori cellulari.
Image Gallery
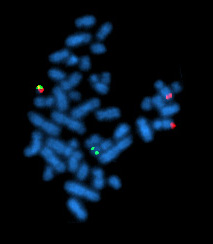
Image Gallery
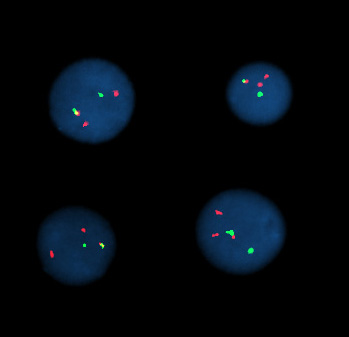
Per i geni codificanti proteine, la localizzazione subcellulare e tissutale della proteina è informativa sulla funzione. Tecniche immunocitochimiche con anticorpi specifici rappresentano un approccio classico, ma richiedono reagenti affidabili e possono non consentire osservazioni dinamiche in tempo reale. Un’alternativa potente consiste nell’impiegare geni reporter, proteine la cui attività è facilmente rilevabile (luminescente, colorimetrica o fluorescente), sotto il controllo degli elementi regolativi del gene di interesse.
Inserendo le regioni promotore/enhancer a monte di un reporter si ottiene un costrutto che riproduce il profilo di espressione del gene nativo, permettendo di visualizzare quando e dove esso è attivo (Figura 04.10-03). Varianti dello stesso schema consentono di dissezionare il contributo di specifiche sequenze cis‑regolative: porzioni mutate o cancellate del promotore rivelano siti necessari o sufficienti alla regolazione (Figura 04.10-03). Reporter comunemente impiegati includono luciferasi, β‑galattosidasi e proteine fluorescenti intrinseche.
Un approccio complementare unisce, in un’unica proteina di fusione, la sequenza codificante del gene in studio e quella di una proteina fluorescente. L’aggiunta della proteina fluorescente all’estremità N‑ o C‑terminale, con adeguato linker, consente di seguire la proteina risultante nelle cellule vive e nei tessuti mediante microscopia a fluorescenza (Figura 04.10-04). In molti casi la proteina di fusione conserva le proprietà della proteina nativa, permettendo di mappare compartimenti subcellulari, traffico intracellulare e dinamiche di assemblaggio di complessi.
L’ampio spettro di varianti delle proteine fluorescenti, con differenti massimi di eccitazione/emissione e caratteristiche fotofisiche, rende possibile marcare simultaneamente più specie proteiche e distinguere segnali in canali multipli (Figura 04.10-05). Strategie come FRET o BiFC, oltre a GFP “destabilizzate” a emivita breve, permettono rispettivamente di studiare interazioni proteina‑proteina e di monitorare variazioni rapide di espressione:
- considerazioni di progetto: scelta della posizione della fusione (N‑ o C‑terminale) e del linker per preservare la funzione; uso di knock‑in al locus endogeno per pattern più fedeli rispetto a espressione da vettori episomali;
- controlli sperimentali: verifica di funzionalità della proteina di fusione mediante saggi di rescate o fenotipici; confronto con localizzazione definita da anticorpi quando disponibili;
- varianti utili: reporter con sequenze di degradazione per aumentare risoluzione temporale; luciferasi per misure quantitative in bulk e in vivo; sistemi split‑reporter per rilevare complessi proteici;
- limiti e precauzioni: possibili artefatti da sovraespressione, interferenze sulla maturazione o sul traffico proteico, fototossicità durante imaging prolungato.
La combinazione di reporter regolativi e fusioni fluorescenti offre un quadro dinamico e spaziale dell’attività genica e proteica, complementare alle misure trascrittomiche. Questi strumenti, accoppiati a manipolazioni genetiche mirate e a sistemi di espressione controllata, consentono di collegare in modo causale elementi cis‑regolativi, livelli di trascrizione, localizzazione proteica e funzioni cellulari, preparando il terreno per la produzione su larga scala delle proteine interessate e la loro caratterizzazione biochimica e strutturale.
Image Gallery
Image Gallery
Image Gallery
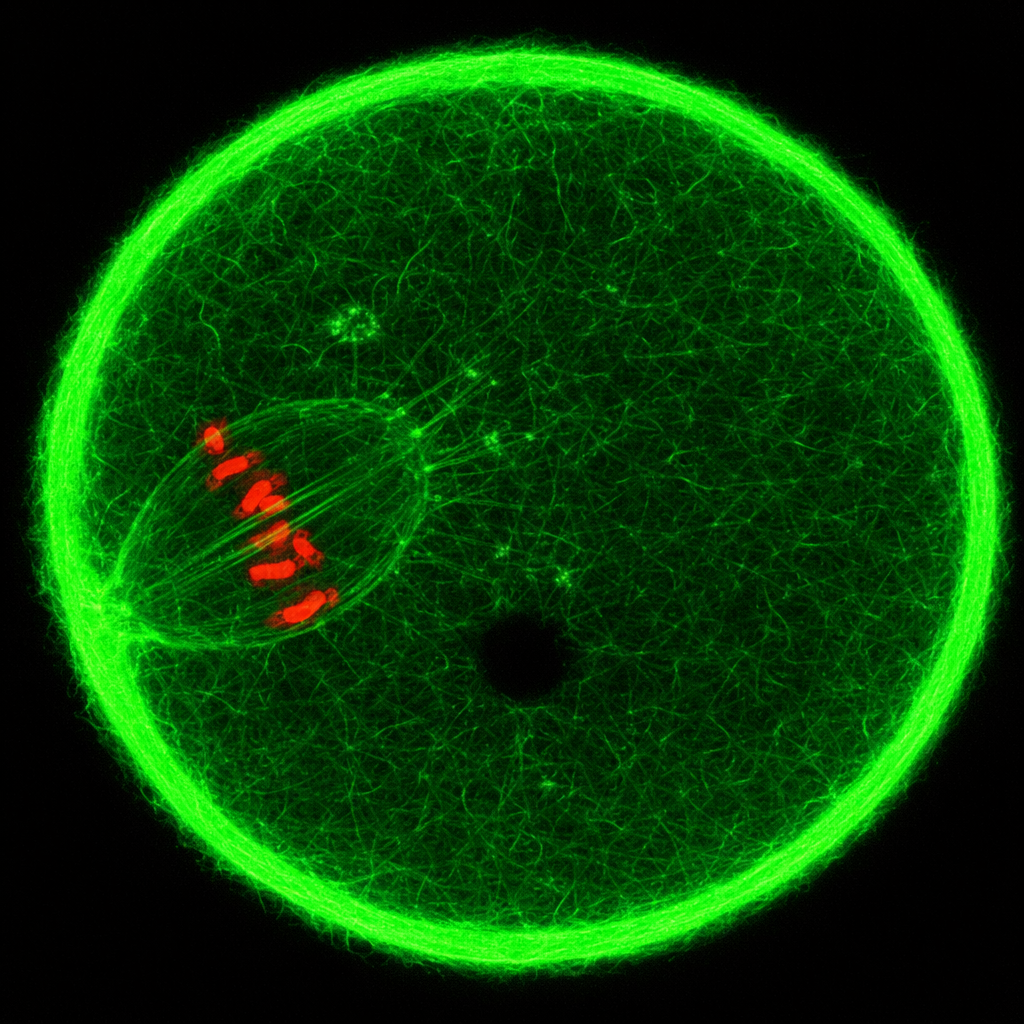
Image Gallery
00084-2/asset/a1168d44-0964-405c-b796-6acce7f26379/main.assets/gr6_lrg.jpg)
Un modo potente per inferire il ruolo di un gene consiste nell’osservare come cambia il fenotipo quando la sua attività viene compromessa. Ben prima dell’era del clonaggio molecolare, la genetica sperimentale si è basata su individui mutanti che emergevano spontaneamente in una popolazione. Gli esemplari con tratti riconoscibili venivano selezionati e studiati attraverso incroci, secondo la logica mendeliana: si pensi, per esempio, a moscerini con ali vestigiali o a lieviti incapaci di sintetizzare un determinato amminoacido. Tramite test di complementazione e analisi di segregazione era possibile inferire se mutazioni diverse coinvolgessero lo stesso gene o vie differenti. Poiché l’insorgenza spontanea di mutazioni è rara, si è fatto largo uso di mutagenesi indotta: radiazioni (UV, raggi X) e agenti chimici come EMS o ENU introducono alterazioni casuali nella sequenza del DNA, generando ampie collezioni di mutanti, ciascuno dei quali può essere analizzato individualmente. Questo approccio, noto come genetica “forward”, segue in genere uno schema ricorrente: mutagenesi, selezione o schermo fenotipico, mappaggio genetico e, oggi, identificazione del gene mediante sequenziamento. L’efficacia di tali strategie dipende da tempi rapidi di riproduzione e dalla possibilità di condurre analisi genetiche controllate. Alcuni organismi modello risultano particolarmente adatti:
- batteri e lieviti, grazie a cicli vitali brevi, facilità di manipolazione e possibilità di selezioni stringenti su terreni di crescita;
- nematodi come Caenorhabditis elegans e insetti come Drosophila melanogaster, per la ricchezza di strumenti genetici (ad esempio cromosomi “balancer” in Drosophila) e la tracciabilità dei caratteri;
- vertebrati quali Danio rerio e Mus musculus, che consentono di esplorare processi complessi di sviluppo e fisiologia, pur richiedendo tempi più lunghi.
Varianti sperimentali, come i mutanti termosensibili o ipomorfi, permettono di attenuare la funzione genica senza abolirla del tutto, facilitando lo studio di geni essenziali. Con l’integrazione delle tecnologie di clonaggio e sequenziamento, oggi i mutanti selezionati possono essere rapidamente collegati alla lesione molecolare responsabile, completando il percorso dal fenotipo al gene con elevata precisione.
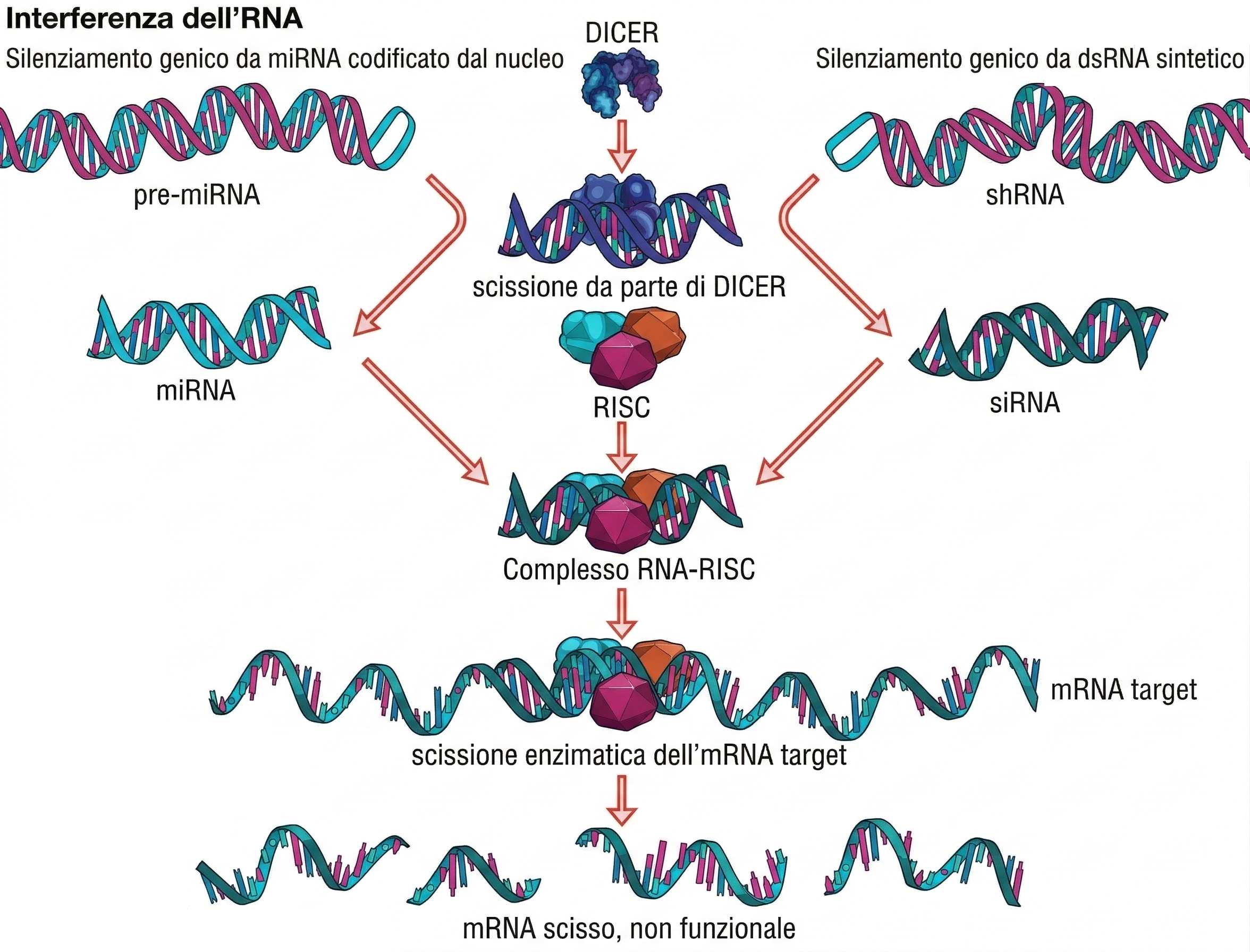
La genetica “reverse” parte da una sequenza nota e ne riduce o elimina l’attività per osservare le conseguenze cellulari o organismiche. Tra gli strumenti più rapidi figura l’interferenza a RNA (RNAi), descritta alla fine degli anni Novanta come risposta endogena di piante e animali contro alcuni virus e elementi mobili. L’introduzione di RNA a doppio filamento (dsRNA) omologo a un trascritto bersaglio attiva un apparato cellulare specializzato: l’enzima Dicer processa il dsRNA in piccoli frammenti, i siRNA, che vengono caricati sul complesso RISC contenente proteine della famiglia Argonaute. La guida a singolo filamento del siRNA dirige RISC verso l’mRNA complementare, inducendone il taglio o la repressione traducionale. In diversi organismi, inclusi C. elegans e molte piante, la risposta può amplificarsi grazie a polimerasi RNA-dipendenti che generano ulteriori siRNA, sostenendo l’inibizione nel tempo e, talora, diffondendola a livello sistemico. Nelle colture cellulari di mammifero si impiegano siRNA sintetici o vettori che esprimono shRNA, evitando l’innesco di risposte antivirali tipiche di dsRNA lunghi. L’applicazione pratica dell’RNAi è flessibile: i dsRNA possono essere veicolati per trasfezione, microiniezione o somministrazione alimentare (in C. elegans), e nei modelli invertebrati la risposta tende a propagarsi nei diversi tessuti. Introdurre un RNA a doppio filamento in C. elegans o nei moscerini della frutta induce il verme o l’insetto così modificato geneticamente in modo da produrre l’RNA che innesca l’RNAi (Figura 04.10-06). Questi RNA vengono trasformati in siRNA, che successivamente si distribuiscono in tutto il corpo dell’animale, inibendo l’espressione del gene bersaglio nei vari tipi di tessuto. Per genomi completamente annotati, è possibile progettare pannelli di siRNA o shRNA per valutare sistematicamente la funzione di interi insiemi di geni. Nonostante la versatilità, l’RNAi presenta criticità: l’effetto è generalmente un “knock-down” parziale e transitorio, con efficienza variabile tra tipi cellulari; possono verificarsi effetti fuori bersaglio dovuti a complementarietà imperfetta o saturazione della via dei piccoli RNA. L’interpretazione rigorosa richiede controlli multipli, come siRNA indipendenti per lo stesso gene, analisi di “rescue” con trascritti refrattari all’RNAi e monitoraggio della specificità molecolare.
Image Gallery
Quando occorre abolire stabilmente l’attività di un gene o introdurre modifiche precise nella sua sequenza, le tecniche di DNA ricombinante consentono l’“editing” mirato del genoma. A differenza dell’RNAi, che riduce temporaneamente l’espressione, il “knock-out” inattiva il gene alla fonte, mentre il “knock-in” sostituisce l’allele endogeno con una versione modificata che altera la funzione proteica o inserisce tag e cassette reporter.
Storicamente, nei mammiferi l’approccio si è basato sulla ricombinazione omologa in cellule staminali embrionali (ES) di topo. Un costrutto di DNA progettato con regioni omologhe al locus bersaglio veicola l’evento di sostituzione: mediante marcatori di selezione positivi/negativi è possibile isolare le cellule correttamente ricombinanti. Queste cellule ES ingegnerizzate vengono quindi introdotte in embrioni precoci per generare animali chimerici e, dopo trasmissione germinale, ceppi mutanti stabili (Figura 04.10-07). Usando una tecnica simile, l’attività di entrambe le copie di un gene può essere soppressa, creando un knock out genico; la disponibilità di librerie mirate ha reso sistematica l’annotazione funzionale del genoma murino (Figura 04.10-08). Gli organismi così modificati sono transgenici o organismi geneticamente modificati, e il segmento introdotto è definito transgene.
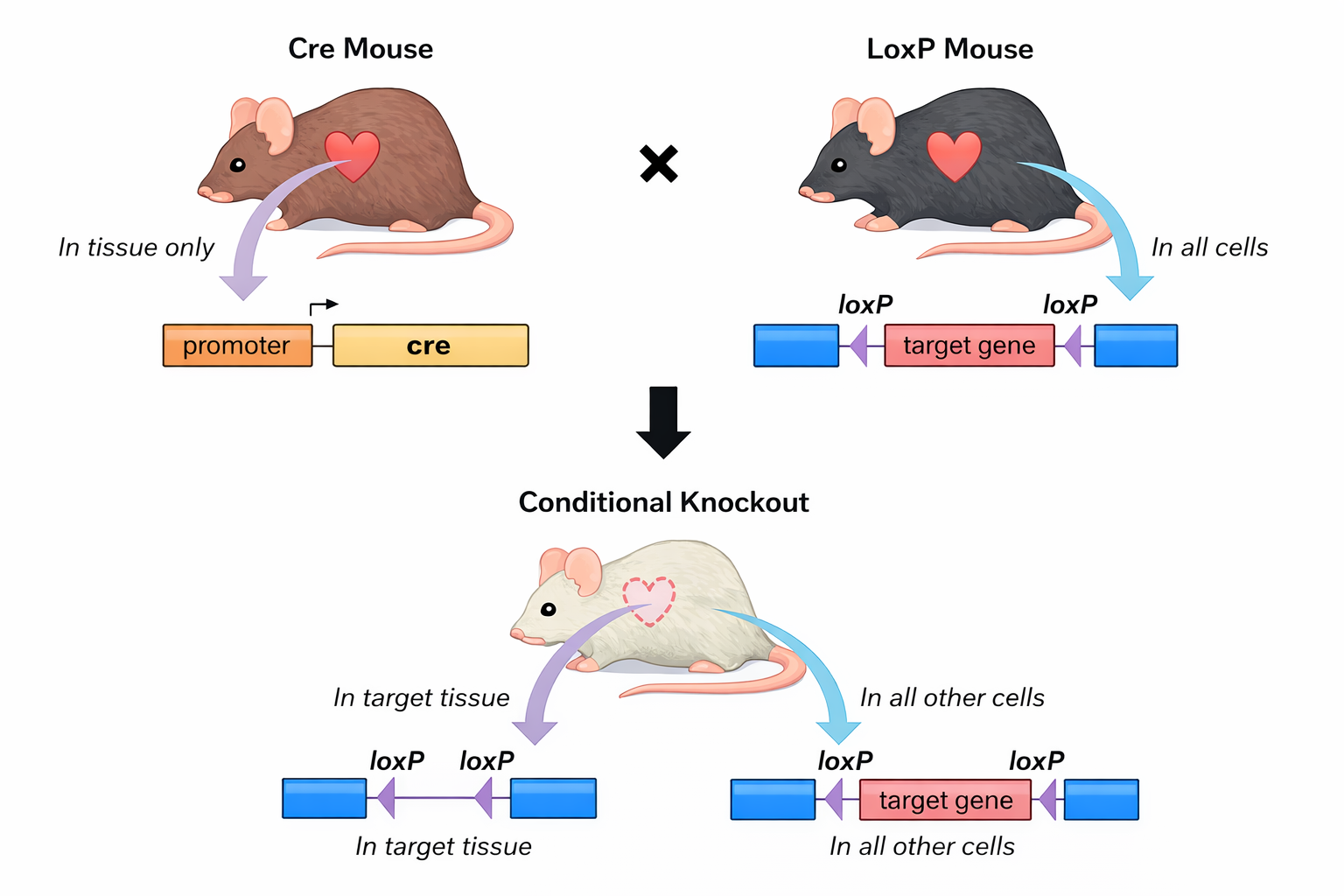
In molti casi la perdita completa di un gene essenziale provoca letalità embrionale, impedendo lo studio di funzioni tardive o tessuto-specifiche. Per ovviare a ciò si impiegano “knock-out” condizionali basati su ricombinasi sito-specifiche. La strategia più diffusa utilizza Cre-loxP: si inseriscono sequenze loxP ai lati di un esone critico (“floxing”) e si esprime la ricombinasi Cre sotto il controllo di promotori ristretti a un tessuto o attivabili farmacologicamente (ad esempio Cre-ERT2). L’enzima riconosce i siti loxP e rimuove la regione interposta, inattivando il gene in modo spaziale e/o temporale (Figura 04.10-09). Sistemi alternativi, come Flp-FRT, ampliano ulteriormente la versatilità combinatoria.
Accanto alla ricombinazione omologa mediata da cellule ES, strumenti di editing di nuova generazione, come la nucleasi CRISPR-Cas9, permettono di introdurre mutazioni mirate direttamente in zigoti o in linee cellulari, accelerando la produzione di knock-out e knock-in e facilitando modifiche puntiformi, delezioni definite o inserzione di cassette reporter. In modelli non murini, inclusi zebrafish e organismi invertebrati, CRISPR ha fortemente ridotto i tempi di generazione delle linee mutanti, mantenendo la possibilità di strategie condizionali quando combinato con sistemi Cre/lox o con guide RNA espresse in modo tessuto-specifico.
La scelta tra “knock-down” tramite RNAi e “knock-out/knock-in” dipende dall’obiettivo sperimentale:
- validazione rapida di un gene e analisi di fenotipi reversibili favoriscono l’RNAi;
- necessità di abolizione completa della funzione, modellazione di varianti patologiche o tracciamento genetico richiedono alterazioni stabili del DNA;
- per geni essenziali o funzioni pleiotropiche, i knock-out condizionali permettono risoluzione spaziale e temporale;
- controlli di specificità, esperimenti di “rescue” e strategie ortogonali (RNAi, CRISPR, alleli condizionali) aumentano la robustezza delle conclusioni.
Insieme, questi approcci costituiscono un repertorio complementare: dall’osservazione fenotipica dei mutanti alla manipolazione precisa del genoma, consentono di collegare in modo causale la sequenza di un gene alle sue funzioni cellulari e organismiche, nonché di generare modelli rigorosi di patologie umane utili alla ricerca preclinica.
Image Gallery
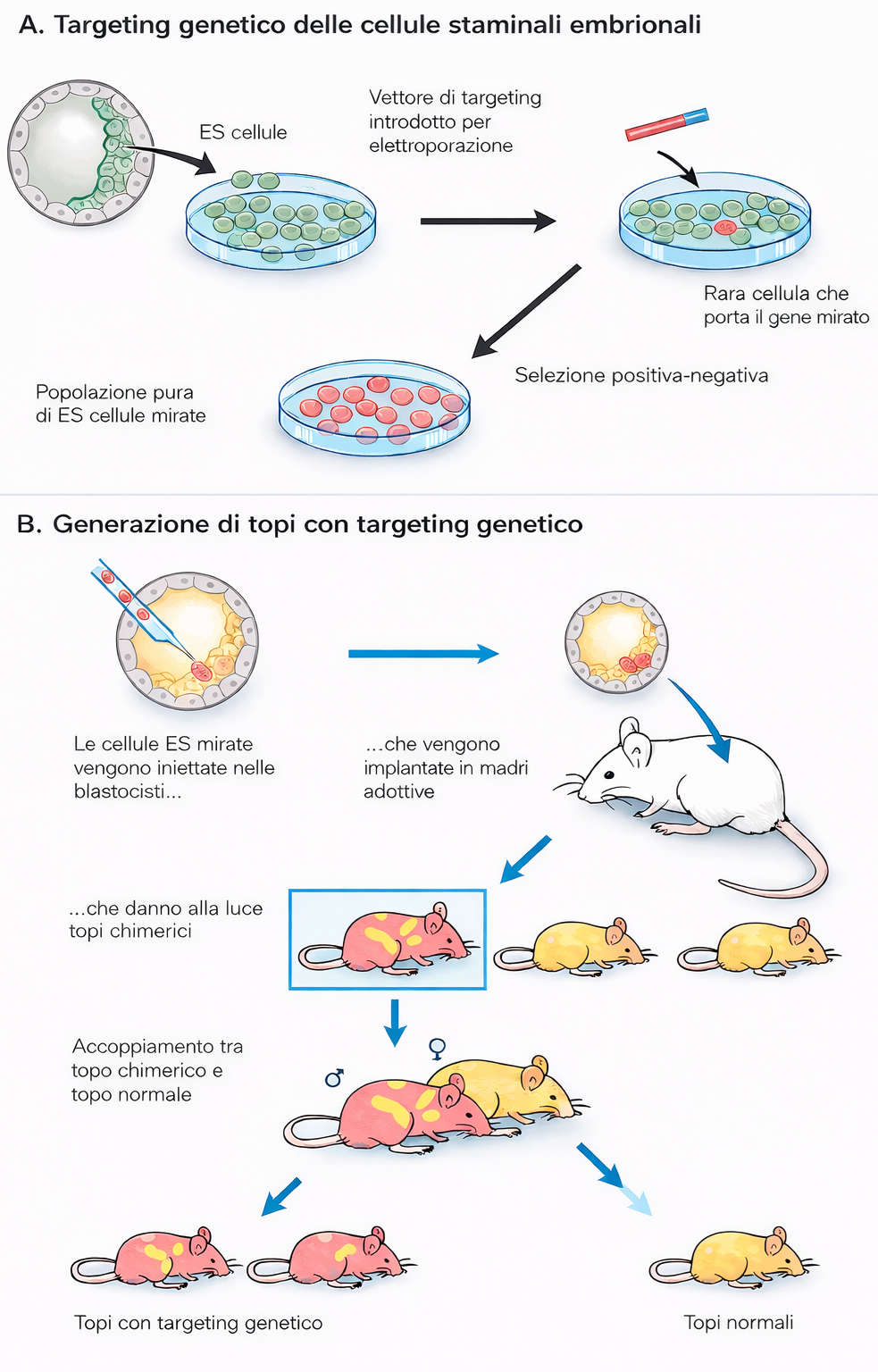
Image Gallery
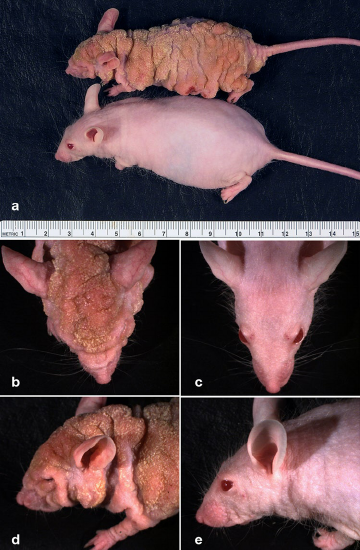
Image Gallery
I procarioti hanno evoluto diversi dispositivi difensivi contro il materiale genetico estraneo, tra cui i ben noti enzimi di restrizione. Un’ulteriore barriera, studiata inizialmente come risposta immunitaria adattativa dei batteri, ha consentito di sviluppare una piattaforma estremamente versatile per la manipolazione del genoma in cellule, tessuti e organismi. Questo sistema, denominato CRISPR, sfrutta nucleasi associate a CRISPR, in particolare Cas9, capaci di introdurre rotture a doppio filamento del DNA. A differenza delle endonucleasi di restrizione, il riconoscimento della sequenza bersaglio non dipende da un sito consensuale proteina-DNA, bensì dall’appaiamento tra una guida a RNA e la regione complementare nel genoma, purché sia presente un breve motivo adiacente denominato PAM (per la SpCas9 tipica, sequenze NGG) (Figura 04.10-10). L’espressione del gene codificante Cas9 è stata resa possibile in un’ampia varietà di sistemi sperimentali; di conseguenza, per indirizzare il taglio su uno o più loci è sufficiente fornire l’RNA guida più adatto.
Le rotture a doppio filamento generate da Cas9 vengono riparate dalle cellule principalmente attraverso due vie: la giunzione non omologa delle estremità (NHEJ), che può generare inserzioni o delezioni con possibili frameshift, e la ricombinazione omologa (HDR), che impiega una sequenza stampo per il ripristino accurato dell’informazione. Pertanto, per sostituire in modo mirato un gene o introdurre varianti definite, si affianca al sistema CRISPR un DNA donatore contenente la modifica desiderata e flanking omologhi, in modo che la rottura sia riparata utilizzando questo stampo e producendo una sostituzione precisa (Figura 04.10-10). L’efficienza, in molte circostanze, è tale da rendere il sistema uno strumento privilegiato per la generazione di organismi transgenici e di linee cellulari ingegnerizzate.
Oltre al taglio catalitico, la piattaforma consente interventi di regolazione genica. Una variante cataliticamente inattiva, dCas9, può essere fusa a domini regolativi: attivatori trascrizionali per incrementare l’espressione o repressori/complessi epigenetici per silenziarla. L’RNA guida indirizza il complesso ibrido al promotore o a regioni regolative del gene bersaglio, permettendo interventi programmabili di attivazione (CRISPRa) o interferenza (CRISPRi) senza alterare la sequenza del DNA (Figura 04.10-10). Sono stati inoltre sviluppati editor di basi e editor “prime”, che combinano dCas9 o Cas9 “nickase” con deaminasi o trascrittasi inversa per introdurre cambi puntuali o piccole riscritture senza rotture a doppio filamento.
La trasposizione del sistema CRISPR dai batteri a numerosi organismi modello, come topo, pesce zebra, nematodi, insetti, riso e grano, ha ridefinito lo studio della funzione genica, analogamente all’impatto storico degli enzimi di restrizione e dell’RNAi. L’adozione di varianti ad alta fedeltà di Cas9 e di nucleasi alternative (per esempio Cas12a) ha ampliato lo spettro di bersagli e ridotto gli effetti off-target, con ricadute su biologia di base, medicina e biotecnologie. Questi progressi, scaturiti da ricerche fondamentali su processi microbici, testimoniano il valore trasformativo della curiosità scientifica non finalizzata.
Image Gallery
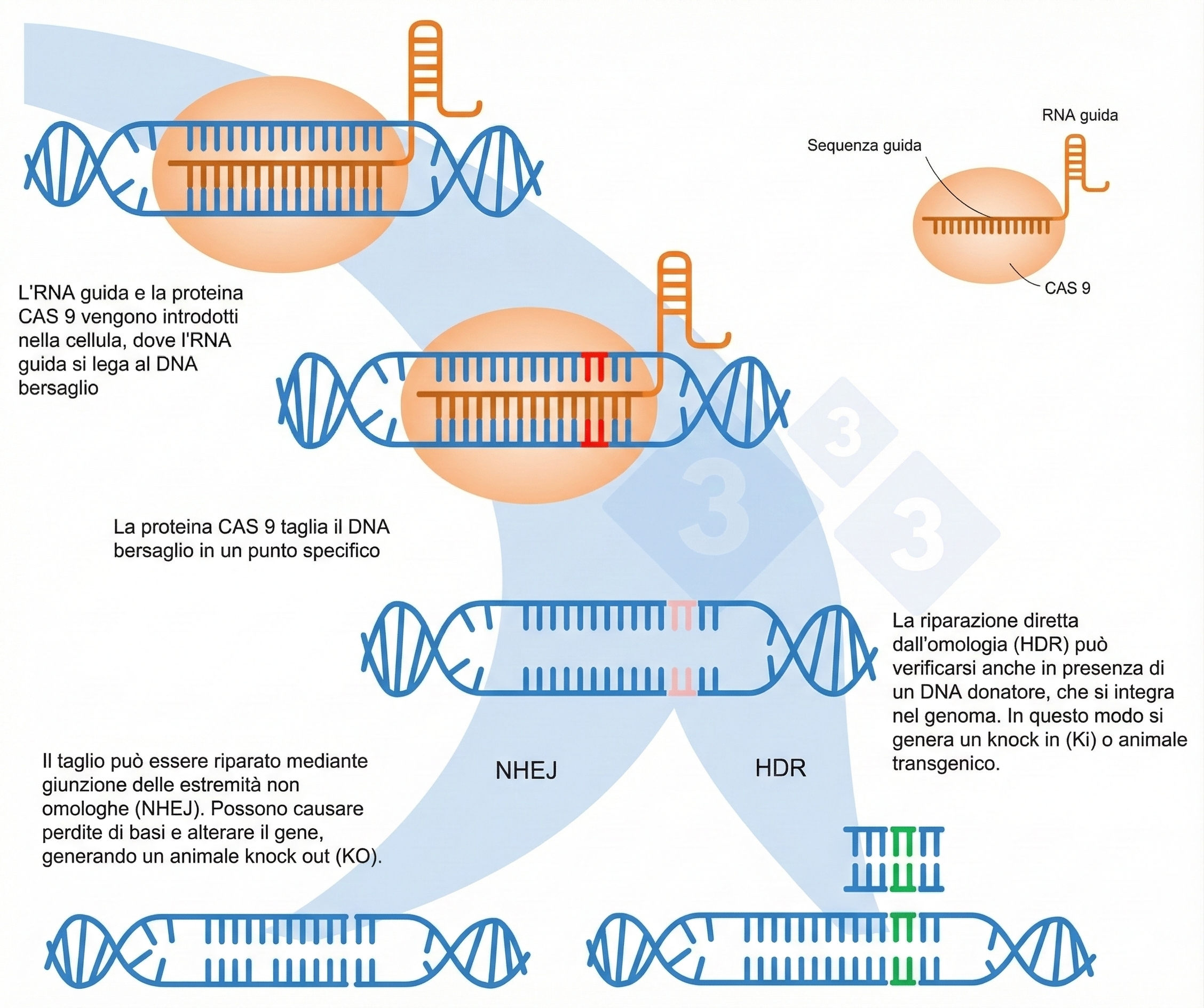
Image Gallery
Image Gallery
Sul piano tecnico, piattaforme transgeniche, incluso CRISPR, renderebbero teoricamente possibile l’editing dei geni della linea germinale umana; tale prospettiva solleva però questioni etiche e regolatorie sostanziali e non è ritenuta accettabile. In ambito sperimentale, invece, le stesse tecnologie sono ampiamente impiegate per creare modelli animali di malattia in cui varianti genetiche patogene svolgono un ruolo fondamentale. L’analisi genomica su larga scala consente di identificare rapidamente mutazioni associate a patologie o a rischio aumentato. Queste alterazioni vengono riprodotte in modelli murini e in altri organismi, generando fenotipi che mimano aspetti clinici e istopatologici della condizione umana. Tali modelli permettono di delineare i meccanismi cellulari e molecolari alla base della malattia e di valutare strategie farmacologiche o genetiche con potenziale traduzione clinica. Un caso esemplare riguarda la distrofia muscolare di Duchenne, causata da mutazioni nel gene della distrofina. I topi mdx, che presentano una mutazione inattivante di distrofina, mostrano degenerazione muscolare e deficit funzionali paragonabili a quelli osservati nei pazienti. Interventi terapeutici in modelli murini, come l’exon skipping mediato da oligonucleotidi antisenso o approcci di editing per ripristinare la cornice di lettura, hanno migliorato la funzione muscolare e la stabilità del sarcolemma, offrendo una prova di principio dell’efficacia di terapie mirate. Questi risultati preclinici hanno contribuito allo sviluppo di trattamenti oggi in uso clinico e guidano il disegno di sperimentazioni di nuova generazione.
Le tecnologie del DNA ricombinante hanno inciso profondamente anche sulla biologia delle piante, che possiedono caratteristiche – in primis la totipotenza cellulare – particolarmente favorevoli alla manipolazione genetica. Coltivando frammenti di tessuti vegetali in condizioni sterili con nutrienti e regolatori di crescita appropriati, alcune cellule vanno incontro a proliferazione indeterminata formando un callo. Regolando accuratamente la composizione del mezzo, si può indurre l’organogenesi e, in diverse specie, rigenerare intere piante da singole cellule del callo. In specie modello come tabacco, petunia, carota, patata e Arabidopsis, cellule coltivate e transfettate con DNA possono dare origine a piante transgeniche (Figura 04.10-11). Questa capacità ha accelerato la dissezione dei processi fondamentali della biologia vegetale, tra cui la percezione dei fitoregolatori e i circuiti di morfogenesi ed espressione genica. Sul versante applicativo, gli strumenti transgenici hanno abilitato tratti agronomici di interesse, per esempio:
- rimodulazione della composizione dei semi, variando il rapporto tra lipidi, amido e proteine;
- resistenza a fitofagi e virus, con riduzione degli input chimici;
- tolleranza a condizioni ambientali sfavorevoli, come suoli salini, allagati o aridi;
- arricchimento nutrizionale mirato di metaboliti benefici.
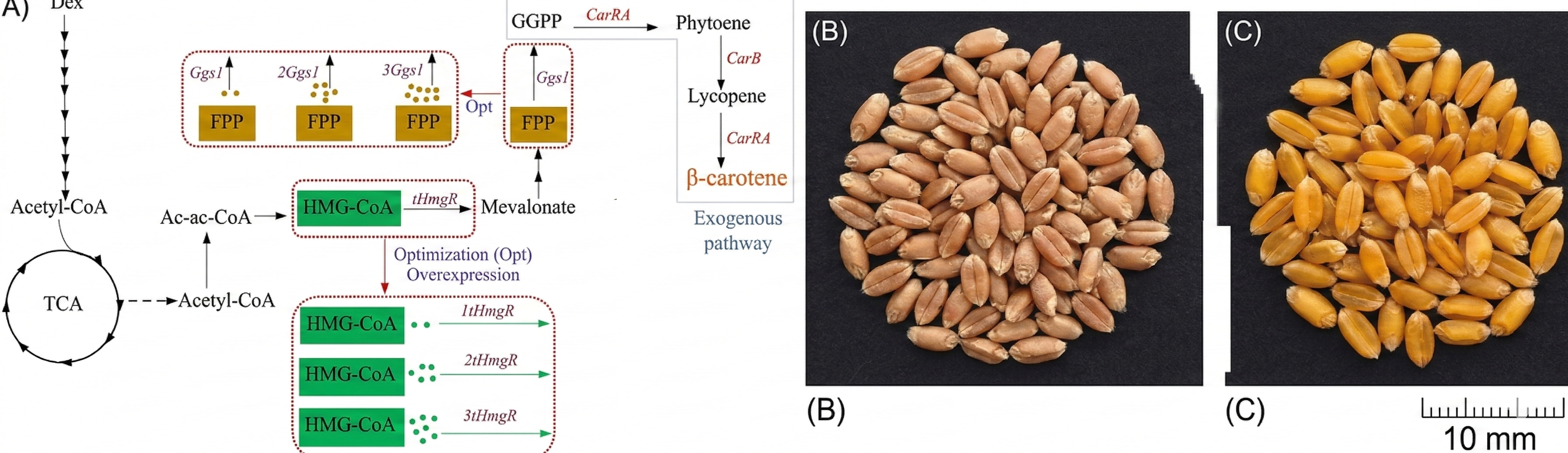
Un esempio di biofortificazione è il “riso dorato”, in cui è stato ricostituito il flusso biosintetico del β-carotene nell’endosperma del chicco (Figura 04.10-12). L’incremento del precursore della vitamina A nei granelli, di colore giallo-arancio, mira a mitigare la carenza di vitamina A in regioni dove il riso costituisce il principale alimento di base. Le valutazioni regolatorie e gli studi di sicurezza hanno accompagnato la diffusione di questa coltura in alcuni contesti geografici, con l’obiettivo di integrare strategie nutrizionali e sanitarie. Sul piano tecnologico, la trasformazione è spesso mediata da Agrobacterium tumefaciens tramite plasmidi Ti ingegnerizzati o mediante biolistica; più recentemente, l’editing CRISPR ha velocizzato la produzione di alleli mirati e l’introduzione di tratti complessi per via cis-genica o intragenica, riducendo inserzioni ectopiche e tempi di ottenimento delle linee.
Image Gallery
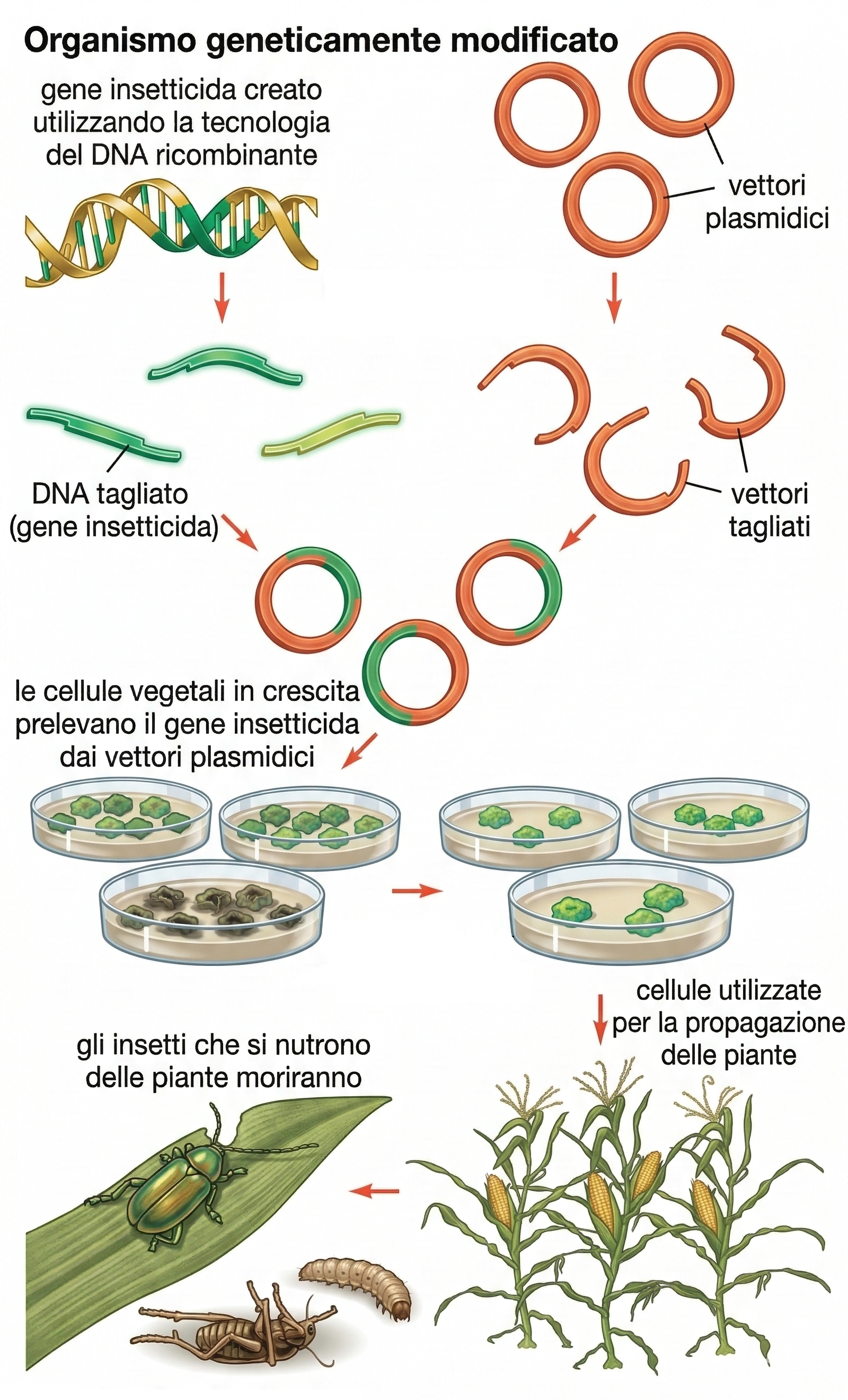
Image Gallery
Un vantaggio cruciale dell’ingegneria genetica è la possibilità di ottenere quantità elevate di proteine, incluse quelle raramente espresse in natura. Ciò si realizza tramite vettori di espressione che incorporano sequenze regolative per una trascrizione e traduzione efficienti del gene inserito. Esistono piattaforme ottimizzate per batteri, lieviti, cellule di insetto e di mammifero, con promotori e segnali di processamento specifici per ciascun sistema (Figura 04.10-13). Poiché il vettore replica durante le divisioni cellulari, le cellule transfettate possono sintetizzare la proteina d’interesse a livelli che arrivano spesso all’1–10% del totale proteico cellulare, facilitandone l’isolamento. L’uso di tag di affinità, segnali di secrezione o domini di solubilità semplifica purificazione e caratterizzazione. La scelta del sistema ospite è guidata dalla necessità di modifiche post-traduzionali appropriate (per esempio glicosilazioni complesse in cellule di mammifero) e da requisiti di ripiegamento, attività e sicurezza. Ottimizzazione di codoni, controllo dell’espressione e condizioni di coltura/fermentazione contribuiscono a massimizzare le rese. Questa flessibilità ha reso accessibili proteine di interesse biomedico – ormoni come l’insulina, fattori di crescita, antigeni vaccinali – e ha permesso studi strutturali e funzionali prima impraticabili, specialmente per recettori di membrana e regolatori trascrizionali a bassa abbondanza. Le tecnologie di DNA ricombinante consentono un passaggio bidirezionale tra informazione genetica e prodotto proteico: dalla proteina al gene e, viceversa, dal gene alla proteina, favorendo l’analisi integrata di struttura, funzione e regolazione su molteplici livelli sperimentali (Figura 04.10-14).
Image Gallery
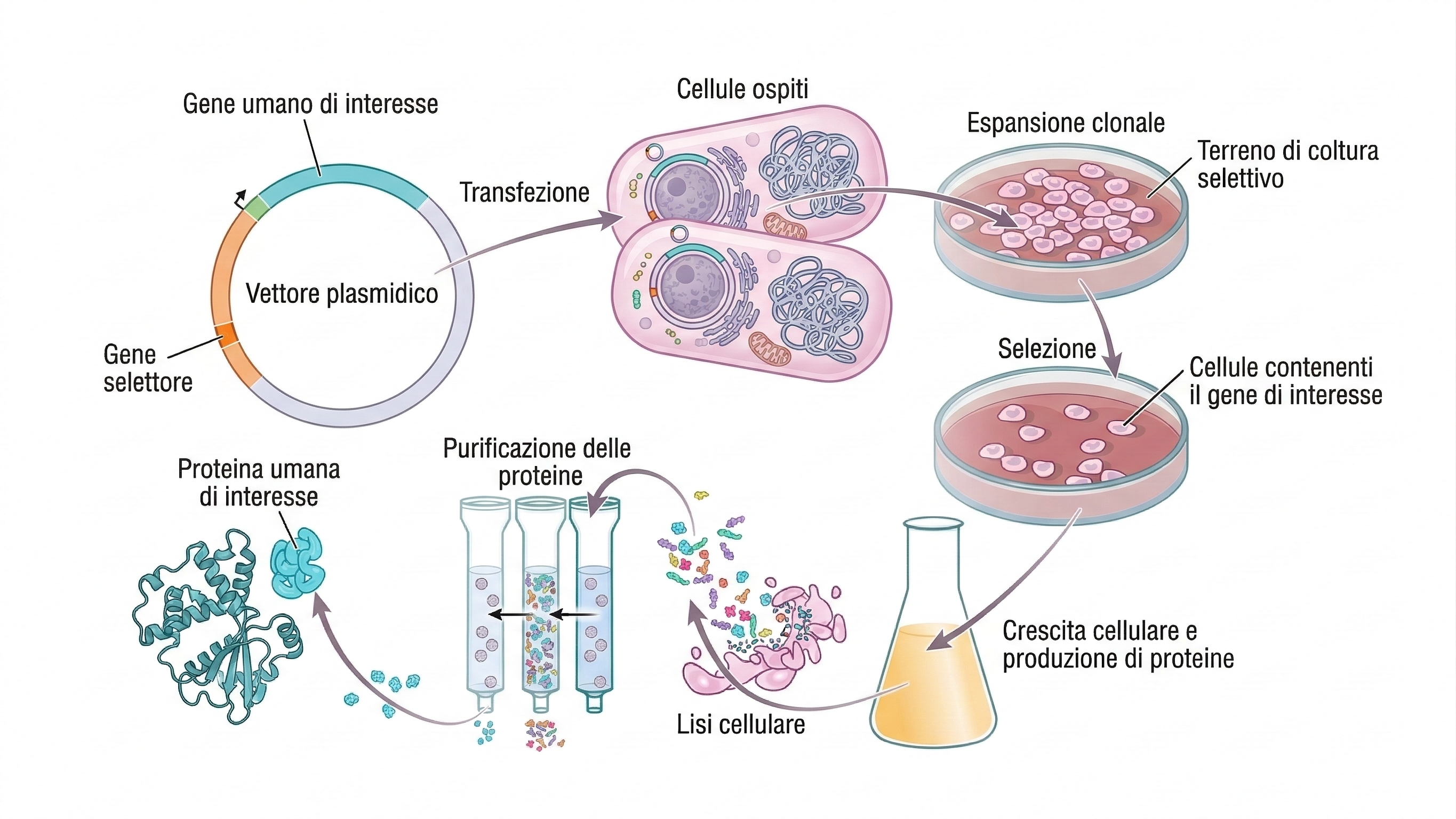
Image Gallery