


La glicolisi ottiene l’energia chimica ossidando il glucosio a piruvato
TOPICS
Definizione

Le sequenze di DNA dei genomi moderni costituiscono un archivio storico dei cambiamenti che hanno sostenuto l’adattamento biologico. Il confronto sistematico tra genomi di organismi appartenenti a lignaggi distanti consente di ricostruire aspetti chiave della storia evolutiva, evidenziando come antiche innovazioni genetiche abbiano predisposto nuove funzioni e strategie di vita. Uno dei risultati più solidi della genomica comparata è l’ampia riconoscibilità di geni omologhi, cioè derivati da un antenato comune, anche tra specie separate da intervalli temporali molto estesi. Omologhi di numerosi geni umani sono individuabili con sicurezza in metazoi come nematodi e insetti, in eucarioti unicellulari quali i lieviti e persino in batteri, sebbene la loro organizzazione genomica e il contesto regolatorio possano divergere profondamente.
Nonostante la linea evolutiva dei vertebrati si sia separata da quella di nematodi e insetti oltre 600 milioni di anni fa, l’analisi comparativa tra i genomi di Caenorhabditis elegans, Drosophila melanogaster e Homo sapiens mostra che circa il 50% dei geni in ciascuna di queste specie presenta omologhi chiaramente identificabili nelle altre. In termini pratici, una quota sostanziale del repertorio genico umano era già rappresentata nell’antenato comune di questi taxa. Il riconoscimento degli omologhi, e la distinzione tra ortologhi (divergenza per speciazione) e paraloghi (duplicazione genica seguita da divergenza funzionale), è fondamentale per inferire relazioni di parentela e per mappare l’evoluzione delle funzioni biologiche.
La rete di corrispondenze geniche, integrata con informazioni sulla disposizione relativa dei geni (sintenia), permette di collocare batteri, animali, piante e funghi in un unico albero della vita, consentendo anche l’inferenza del contenuto genico ancestrale per nodi profondi. In questa prospettiva, i genomi non solo raccontano la storia delle specie, ma offrono ipotesi testabili sul modo in cui l’innovazione genetica si è stratificata nel tempo.
Spesso si immagina l’evoluzione come un percorso lineare verso forme “migliori”, ma al livello molecolare la dinamica è il risultato congiunto di mutazione, deriva genetica e selezione naturale. Consideriamo una mutazione puntiforme insorta nella linea germinale: nella maggioranza dei casi l’effetto è neutro o deleterio; più raramente la mutazione è vantaggiosa e aumenta la fitness del portatore. La probabilità di fissazione di una variante dipende dal suo effetto selettivo e dalla dimensione effettiva della popolazione, Ne. Per un allele strettamente neutro, la probabilità di fissazione è \(1/(2N_e)\), mentre la sua velocità di sostituzione a livello di specie, sotto neutralità, eguaglia la velocità di mutazione per sito, \(k = \mu\). Per alleli vantaggiosi di piccolo effetto, la probabilità di fissazione cresce approssimativamente con il coefficiente selettivo \(s\) (in condizioni addittive, ≈ \(2s\) in popolazioni grandi), il che rende più probabile la loro conservazione nel tempo.
Le varianti deleterie sono generalmente eliminate dalla selezione purificante, sebbene possano persistere transitoriamente per deriva, soprattutto in popolazioni di piccole dimensioni. Mutazioni selettivamente neutre o quasi neutre possono fissarsi per deriva, in particolare in regioni del genoma prive di funzione codificante o regolativa evidente. Tuttavia, l’apparente “neutralità” dipende dal contesto: elementi regolatori, sequenze non codificanti con funzione strutturale o siti di legame per proteine possono essere soggetti a vincoli, riducendo il tasso di sostituzione rispetto a regioni realmente prive di funzione.
Nel lungo periodo, l’alternarsi di innovazione mutazionale e filtraggio selettivo produce genomi nei quali alcune porzioni evolvono rapidamente, mentre altre risultano fortemente vincolate. I geni indispensabili per processi cellulari di base — come le RNA e DNA polimerasi, i ribosomi, componenti del metabolismo energetico — mostrano una conservazione elevata tra i taxa. La selezione purificante limita l’accumulo di sostituzioni amminoacidiche in tali proteine, riflettendosi in rapporti \(d_N/d_S < 1\), dove \(d_N\) è il tasso di sostituzione non sinonima e \(d_S\) quello sinonimo. All’opposto, geni coinvolti nell’interazione ospite-patogeno, nella percezione sensoriale o nell’adattamento a nicchie specifiche possono mostrare accelerazioni locali (\(d_N/d_S > 1\)) indicatrici di selezione positiva:
- Deriva e collegamento fisico: varianti neutre possono aumentare di frequenza per autostop genetico quando legate a mutazioni vantaggiose, o essere rimosse per “background selection” in regioni con mutazioni deleterie ricorrenti;
- Struttura del genoma: regioni a bassa ricombinazione, come centromeri e telomeri, amplificano gli effetti del collegamento, alterando i profili di variabilità;
- Vincoli funzionali: elementi essenziali conservano la sequenza; regioni senza funzione rilevante tendono ad accumulare sostituzioni secondo il tasso mutazionale locale.
In sintesi, l’eterogeneità del paesaggio selettivo spiega perché alcuni tratti genomici siano rimasti riconoscibili per 3,5 miliardi di anni, mentre altri si rinnovano rapidamente, fornendo la materia prima per l’innovazione.
Tra specie strettamente imparentate, le differenze che meglio informano sulle tempistiche di divergenza sono spesso quelle quasi neutre, le quali si accumulano con andamento prossimo a un orologio. Se il tasso di sostituzione neutra per sito è \(\mu\), la distanza media per sito tra due linee che divergono da tempo \(t\) è approssimativamente \(D \approx 2\mu t\). Tale proprietà consente di stimare i tempi di separazione tra specie calibrando l’orologio con fossili affidabili o eventi geologici databili, e di costruire alberi filogenetici coerenti con i dati molecolari e paleontologici.
Il confronto dei genomi dei primati superiori (Figura 04.03-01) evidenzia che lo scimpanzé è la specie sorella dell’uomo: l’insieme dei geni è in larga misura condiviso e l’ordine relativo dei loci risulta ampiamente conservato (sintenia estesa). La più vistosa differenza cromosomica è il cromosoma 2 umano, originato dalla fusione testa-testa di due cromosomi ancestrali omologhi a coppie distinte presenti in scimpanzé, gorilla e orangutan; la presenza di sequenze telomeriche interne e di un centromero vestigiale nel cromosoma 2 supporta l’evento di fusione. Oltre a questa riorganizzazione, i genomi umano e dello scimpanzé presentano inversioni e piccole traslocazioni, ma nel complesso le variazioni strutturali sono contenute rispetto alla scala del genoma.
Le analisi di sintenia e delle sequenze consentono persino di inferire l’assetto del genoma del progenitore comune di Homo e Pan, ricostruendo l’ordine dei geni e gli eventi di riarrangiamento avvenuti dopo la separazione delle linee (Figura 04.03-02). Nonostante fenomeni come l’incomplete lineage sorting possano produrre discordanze tra singoli geni, l’insieme delle evidenze colloca stabilmente uomo e scimpanzé come cladi fratelli.
Gli elementi genetici mobili offrono marcatori informativi aggiuntivi. Nel genoma umano è presente circa un milione di copie della famiglia Alu; un numero comparabile si osserva nello scimpanzé, e oltre il 99% di tali elementi occupa posizioni ortologhe nei due genomi. Ciò indica che la grande maggioranza delle inserzioni Alu predata la divergenza uomo–scimpanzé, mentre solo una frazione minore deriva da inserzioni lineage-specific. Analoghe considerazioni valgono per altre famiglie di retrotrasposoni, che possono fungere da “sinapomorfie molecolari” per definire rami filogenetici.
Nel quadro più generale, l’analisi dei cambiamenti nucleotidici e dell’organizzazione genomica fornisce strumenti complementari per ricostruire la storia delle specie:
- sequenze codificanti e segnali proteici aiutano a valutare vincoli funzionali e episodi di selezione positiva mediante indicatori come \(d_N/d_S\);
- regionI presumibilmente neutre (introni, siti sinonimi, intergeniche prive di annotazione funzionale) sono adatte per stimare tassi di sostituzione e calibrare l’orologio molecolare;
- blocchi di sintenia e anomalie cromosomiche (fusioni, inversioni, traslocazioni) informano sugli eventi di riarrangiamento e sulla macroevoluzione del cariotipo;
- inserzioni di elementi mobili, quando condivise in posizioni identiche, fungono da caratteri quasi omoplasi-free per inferenze filogenetiche robuste.
Combinando queste linee di evidenza, il confronto genomico consente di posizionare con precisione specie strettamente correlate nell’albero della vita, di datare la separazione delle loro linee e di inferire lo stato ancestrale dei loci, chiarendo la sequenza degli eventi che hanno condotto agli assetti genomici attuali descritti nella (Figura 04.03-01) e ricostruiti nella (Figura 04.03-02).
Image Gallery
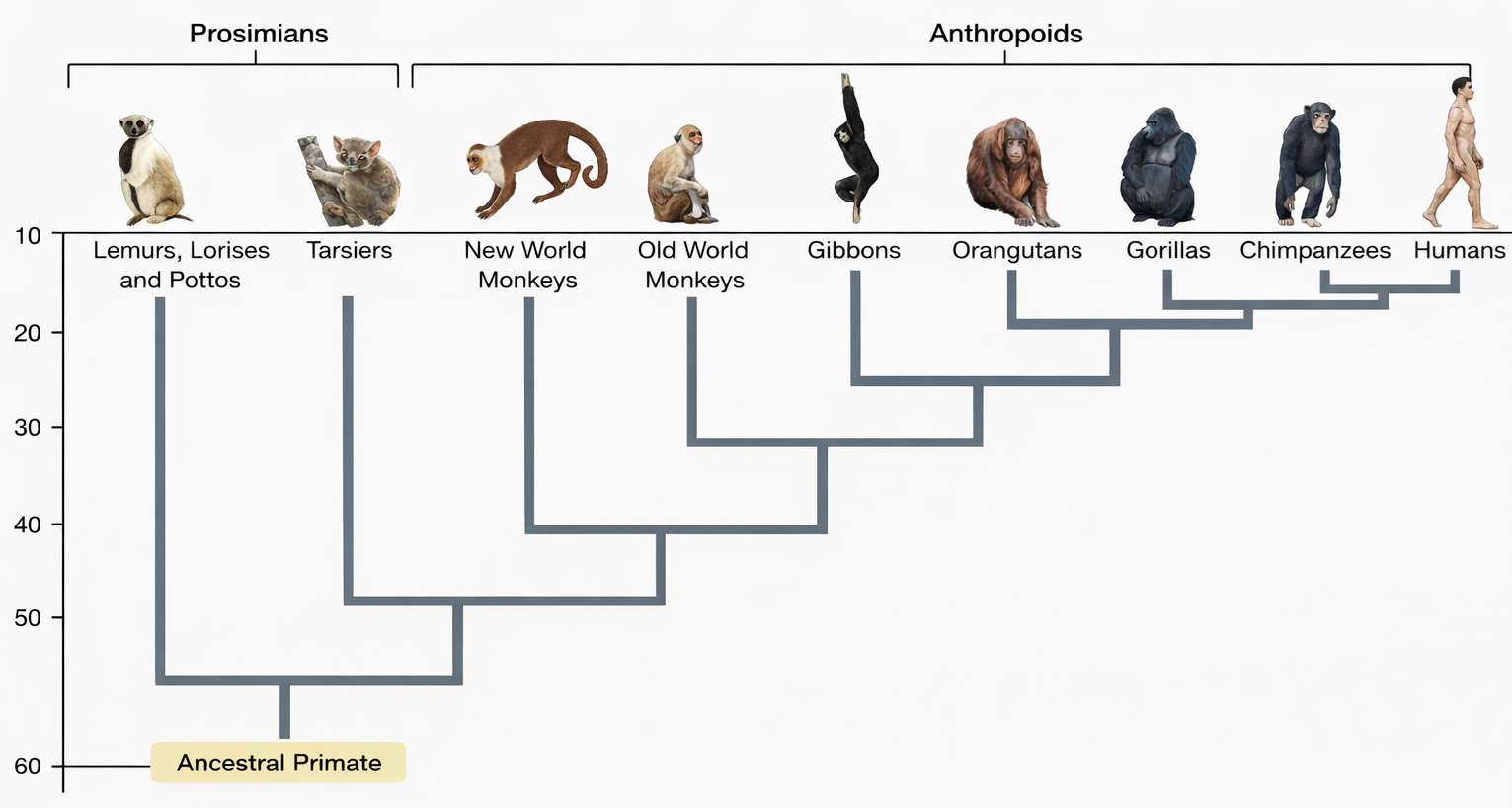
Image Gallery
Image Gallery
Image Gallery
Quando il genoma umano viene messo a confronto con quello di specie più distanti, l’impronta dell’evoluzione diventa più marcata. La linea che conduce a Homo sapiens si è separata da quella del topo circa 75 milioni di anni fa, e i due genomi, pur simili per dimensioni e per numero di geni, hanno seguito traiettorie indipendenti per quanto riguarda la dinamica degli elementi genetici mobili. Sebbene le famiglie di trasposoni e retrotrasposoni condividano affinità di sequenza, la loro distribuzione è il prodotto di proliferazioni e spostamenti avvenuti in ciascun lignaggio dopo la separazione evolutiva (Figura 04.03-03). A questa divergenza contribuisce anche un’intensa riorganizzazione cromosomica: si stima che nell’arco degli ultimi 75 milioni di anni si siano verificati circa 180 eventi di rottura e ri-giunzione che hanno ridisegnato i cromosomi. Un riflesso macroscopico di tali ristrutturazioni è la posizione dei centromeri, per lo più metacentrici o submetacentrici nell’uomo, ma frequentemente telocentrici nel topo. Nonostante il rimaneggiamento, ampi blocchi di sintenia sono rimasti riconoscibili, con geni omologhi conservati nello stesso ordine relativo: oltre il 90% dei genomi umano e murino può essere segmentato in regioni corrispondenti di sintenia conservata.
All’interno di questi blocchi, l’allineamento fine delle sequenze evidenzia che, dalla divergenza, circa il 50% dei nucleotidi ha subito sostituzioni. Su questo sfondo di cambiamento diffuso emergono, però, “isole” di elevata somiglianza tra uomo e topo (Figura 04.03-04), in cui le alterazioni sono state sistematicamente eliminate dalla selezione purificatrice, ossia dalla rimozione delle varianti che riducono la sopravvivenza o la fecondità dei portatori. In termini popolazionistici, una mutazione deleteria con coefficiente di selezione s viene efficacemente eliminata quando \(2N_e s \gg 1\), dove \(N_e\) è la dimensione efficace della popolazione. In pratica, la conservazione di sequenza segnala funzioni biologiche che tollerano poco o nulla il cambiamento.
La potenza inferenziale aumenta includendo ulteriori genomi di mammiferi (ad esempio ratto, cane) e di altri vertebrati, sfruttando un “esperimento naturale” che dura da decine di milioni di anni. Questi confronti mostrano che circa il 4,5% del genoma umano è costituito da DNA conservato in molti altri mammiferi (Figura 04.03-05), ma solo una frazione, circa un terzo, corrisponde a regioni codificanti proteine. La parte restante comprende:
- sequenze regolatrici di espressione genica, come promotori, potenziatori e isolatori;
- RNA non codificanti funzionali, inclusi lncRNA e piccoli RNA coinvolti in controllo post-trascrizionale;
- elementi ultraconservati, tratti di centinaia di basi quasi invariati tra mammiferi, spesso con ruoli regolatori;
- regioni la cui funzione è ancora ignota, ma la cui conservazione suggerisce vincoli selettivi stringenti.
La distinzione tra vincolo selettivo e neutralità può essere formalizzata confrontando la divergenza osservata con l’aspettativa neutrale stimata in siti presumibilmente neutrali (per esempio siti sinonimi a quattro vie); un eccesso di identità rispetto all’atteso indica selezione purificatrice. In modelli semplici, la distanza corretta tra sequenze si può approssimare con il modello di Jukes–Cantor, \(d = -\tfrac{3}{4}\ln\!\bigl(1 - \tfrac{4}{3}p\bigr)\), dove p è la frazione di differenze osservate: scarti negativi rispetto al valore neutrale segnalano conservazione. La rapida diminuzione dei costi di sequenziamento e la disponibilità di allineamenti multispecie consentono di mappare queste regioni con risoluzione crescente, facendo emergere funzioni regolatorie non ancora catalogate.
Image Gallery
Image Gallery
Image Gallery

Spingendo il confronto più indietro nel tempo, la separazione tra pesci e mammiferi risale a circa 400 milioni di anni fa. Su queste scale, la somiglianza di sequenza a livello nucleotidico tende a dissolversi per effetto di sostituzioni casuali e pressioni selettive divergenti, tranne nei tratti soggetti a forte vincolo purificatore. Ciononostante, nel genoma dei pesci è possibile riconoscere la gran parte dei geni umani e, in molti casi, anche elementi regolativi. La numerosità delle famiglie geniche differisce frequentemente tra linee evolutive, in parte per eventi di duplicazione specifici, inclusa una duplicazione dell’intero genoma che ha interessato i teleostei, con conseguente mantenimento o perdita differenziale dei duplicati.
Nonostante un numero di geni relativamente stabile, la dimensione dei genomi vertebrati varia in modo considerevole. Umano, cane e topo presentano genomi nell’ordine di 3 × 10⁹ paia di basi, mentre quello del pollo misura circa un terzo di tale valore. Un caso estremo è il pesce palla, Fugu rubripes (Figura 04.03-06), il cui genoma è vicino a un ottavo di quello dei mammiferi. La compattezza di Fugu riflette introni molto brevi e una ridotta presenza di DNA ripetitivo, pur mantenendo, in larga misura, la posizione degli introni rispetto ai geni omologhi dei mammiferi. Ciò indica che l’architettura exon–intron era già definita nell’antenato comune dei pesci e dei mammiferi e si è conservata strutturalmente.
Quali processi spiegano tali differenze di scala? Analisi comparative su ampi insiemi di genomi hanno rivelato un turnover inatteso di piccoli segmenti di DNA: inserzioni e delezioni si accumulano a tassi elevati, con differenze tra lignaggi. In alcune linee, come quella di Fugu, prevale un bias verso la delezione, che nel lungo periodo determina un’erosione netta delle porzioni non essenziali del genoma. Questa dinamica può essere riassunta come: \[ \Delta L = \lambda_{\text{ins}}\cdot \bar{l}_{\text{ins}} - \lambda_{\text{del}}\cdot \bar{l}_{\text{del}}, \] dove \(\Delta L\) è la variazione attesa di lunghezza del genoma per unità di tempo, \(\lambda\) e \(\bar{l}\) sono rispettivamente tassi e lunghezze medie di inserzioni e delezioni. Se \(\Delta L < 0\), il genoma si “snellisce”. Meccanismi molecolari che alimentano questo bilancio includono:
- espansioni episodiche di elementi trasponibili (retrotrasposizioni e trasposizioni a DNA) e successiva rimozione per ricombinazione omologa non allelica;
- crossing-over ineguale e riparazione di rotture a doppio filamento con microdelezioni;
- selezione contro carichi ripetitivi in specie con grandi popolazioni efficaci, che rende più efficiente la rimozione del DNA non funzionale;
- deriva genetica e vincoli funzionali che modulano il destino dei duplicati genici.
In questo senso, la “versione compatta” del genoma di Fugu rappresenta un utile riferimento: potando gran parte del DNA non essenziale, l’evoluzione ha messo in risalto regioni presumibilmente funzionali, facilitando l’annotazione comparativa.
Image Gallery

Quando il confronto si estende a organismi filogeneticamente molto distanti — oltre primati, roditori, pesci, insetti, nematodi, piante e lieviti, fino ai batteri — le corrispondenze di sequenza con il genoma umano si fanno rare. Eppure, un nucleo di geni essenziali è rimasto riconoscibile grazie alla selezione purificatrice. Tra questi, spicca il gene dell’RNA ribosomiale della subunità minore (SSU rRNA), componente chiave del macchinario di traduzione, la cui struttura è rimasta altamente conservata sin dalle prime fasi della vita sulla Terra (Figura 04.03-07). La misura della divergenza nelle sequenze dell’SSU rRNA tra organismi differenti consente di stimare relazioni filogenetiche profonde, fornendo un tracciato quantitativo della storia evolutiva.
L’uso sistematico dell’SSU rRNA ha permesso di costruire un albero della vita coerente e onnicomprensivo. Pur confermando aspetti della tassonomia morfologica tradizionale, questo approccio ha riservato sorprese: una parte di organismi in precedenza inclusi tra i “batteri” si è rivelata appartenere a un clade distinto, gli archei, separato dai batteri quanto lo sono gli eucarioti dai procarioti tipici. Ne deriva l’attuale ripartizione del vivente in tre domini principali: batteri, archei ed eucarioti (Figura 04.03-08). La filogenomica moderna integra questi marcatori con insiemi di geni “core” e considera anche il ruolo del trasferimento genico orizzontale, particolarmente pervasivo nei procarioti, che può offuscare i segnali per alcuni loci senza però cancellare la traccia evolutiva nei geni maggiormente vincolati.
La diversità genetica del pianeta è in gran parte microbica e invisibile a occhio nudo. Molte specie non sono coltivabili in condizioni standard di laboratorio; la loro scoperta e classificazione si fondano sull’analisi di sequenze direttamente prelevate dall’ambiente, includendo marcatori rRNA e approcci metagenomici su larga scala. Queste strategie rivelano che i microrganismi costituiscono una quota rilevante della biomassa terrestre e svolgono funzioni ecologiche cruciali, spesso passate inosservate se non in occasione di patologie o degrado dei materiali. L’acquisizione massiva di dati di sequenza, provenienti da habitat molto diversi, sta così restituendo un quadro più fedele e meno antropocentrico della biosfera.
Image Gallery
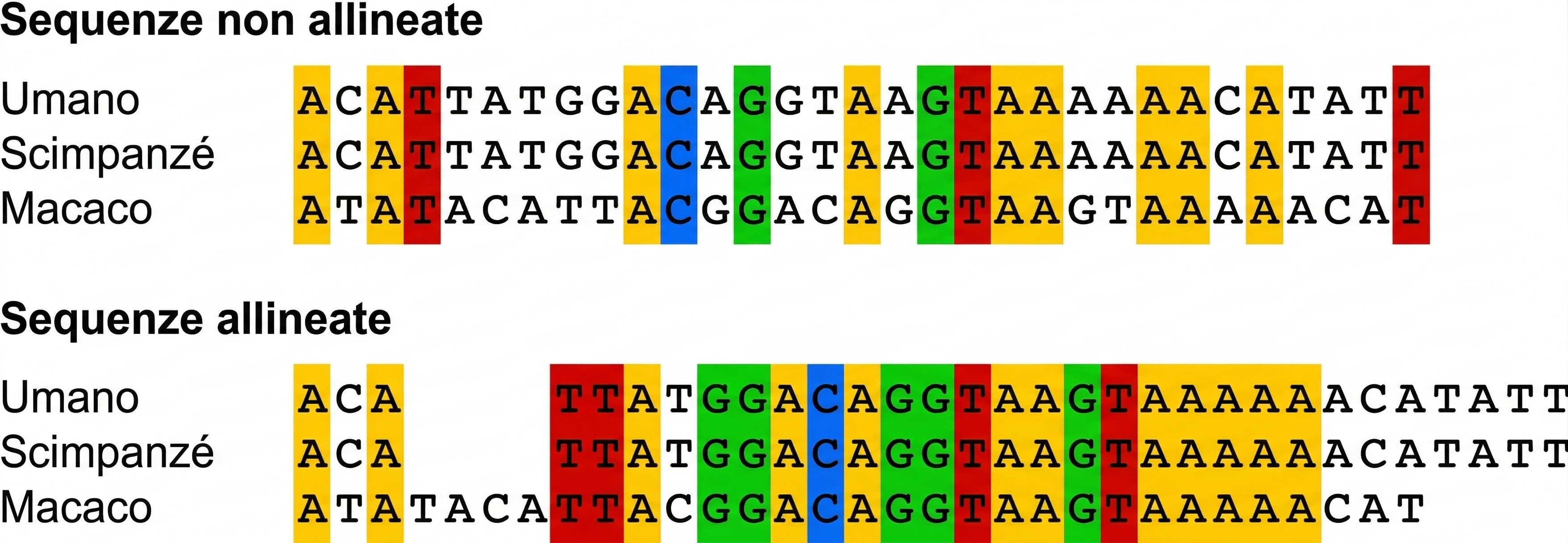
Image Gallery



