


Gli organismi sono sensibili a condizioni di acidità o basicità
TOPICS
Definizione

Il funzionamento degli organismi viventi è sostenuto da reti di reazioni chimiche altamente specifiche che avvengono all’interno delle cellule. La maggior parte di tali trasformazioni è catalizzata da proteine specializzate, gli enzimi, i quali, come qualunque sistema fisico, sono soggetti alla seconda legge della termodinamica. Gli enzimi accelerano reazioni termodinamicamente favorite, ossia processi che incrementano il disordine globale, ma non possono rendere spontanee reazioni che sono sfavorite dal punto di vista energetico. Eppure le cellule devono sintetizzare strutture ordinate e molecole ad alta energia a partire da precursori semplici, operazione che richiede apporto di energia. Per comprendere come gli enzimi rendano possibili e rapide le reazioni chimiche necessarie alla vita, occorre innanzitutto analizzare il bilancio energetico delle molecole: la loro energia libera condiziona la direzione e l’estensione delle trasformazioni chimiche, mentre le variazioni di energia libera misurano l’impatto della reazione sul disordine complessivo dell’Universo. In questo quadro, gli enzimi abbassano la barriera di attivazione che ostacola l’avvio delle reazioni e sfruttano l’accoppiamento fra processi con diversa variazione di energia libera per guidare trasformazioni altrimenti sfavorite, permettendo la costruzione dell’ordine biologico. Senza questa catalisi altamente regolata, i processi cellulari non risulterebbero compatibili con le condizioni della vita.
L’ossidazione del ferro a ruggine ne è un esempio comune: ferro + O₂ → ossidi di ferro + calore. La reazione libera calore e procede in modo sostanzialmente irreversibile nelle condizioni ambientali ordinarie; non si osserva spontaneamente la ricostituzione del metallo lucido dalla ruggine con assorbimento di calore dall’ambiente. In termini termodinamici, la trasformazione disperde energia utilizzabile sotto forma di agitazione termica non più sfruttabile per compiere lavoro, ossia riduce l’energia libera del sistema. Tale dissipazione riflette la perdita di ordine associata alla disposizione iniziale della materia e all’energia chimica immagazzinata nei legami del ferro metallico. Il principio generale che se ne ricava è intuitivo: le reazioni chimiche avanzano nella direzione che comporta una diminuzione dell’energia libera. In altre parole, la spontaneità corrisponde a un “pendio energetico” discendente; si dirà quindi che un processo è favorito dal punto di vista energetico quando si verifica questa discesa.
Anche quando lo stato finale è più stabile dal punto di vista energetico, molte sostanze rimangono cineticamente inerti a temperatura ambiente perché separate dallo stato finale da una barriera di energia, l’energia di attivazione \(E_a\). Per esempio, i vapori di una miscela combustibile richiedono una scintilla per innescare la reazione di combustione, ossia un apporto di energia che consenta alle molecole di superare la barriera e raggiungere lo stato di transizione (Figura 01.13-01). Le cellule non possono semplicemente alzare la temperatura per innescare le loro reazioni: si affidano agli enzimi, che legano in modo selettivo uno o più substrati, li orientano nello spazio, stabilizzano lo stato di transizione e riducono \(E_a\), accelerando così in modo drastico la velocità della reazione specifica (Figura 01.13-01); (Figura 01.13-02); (Figura 01.13-03).
Gli enzimi rientrano nella più ampia classe dei catalizzatori. Una riduzione di \(E_a\) aumenta la frazione di molecole che, alla temperatura della cellula, possiedono energia sufficiente per superare la barriera, come suggerito dalla distribuzione di Maxwell-Boltzmann (Figura 01.13-02). Effetti di accelerazione anche di \(10^{14}\) volte sono documentati per alcuni sistemi, rendendo accessibili, in tempi biologicamente utili, reazioni che a temperatura fisiologica risulterebbero di fatto impercettibili. A titolo esemplificativo, l’anidrasi carbonica accelera l’idratazione della CO₂ di molti ordini di grandezza, mentre la triose-fosfato isomerasi rende rapida un’isomerizzazione altrimenti lenta nelle condizioni cellulari.
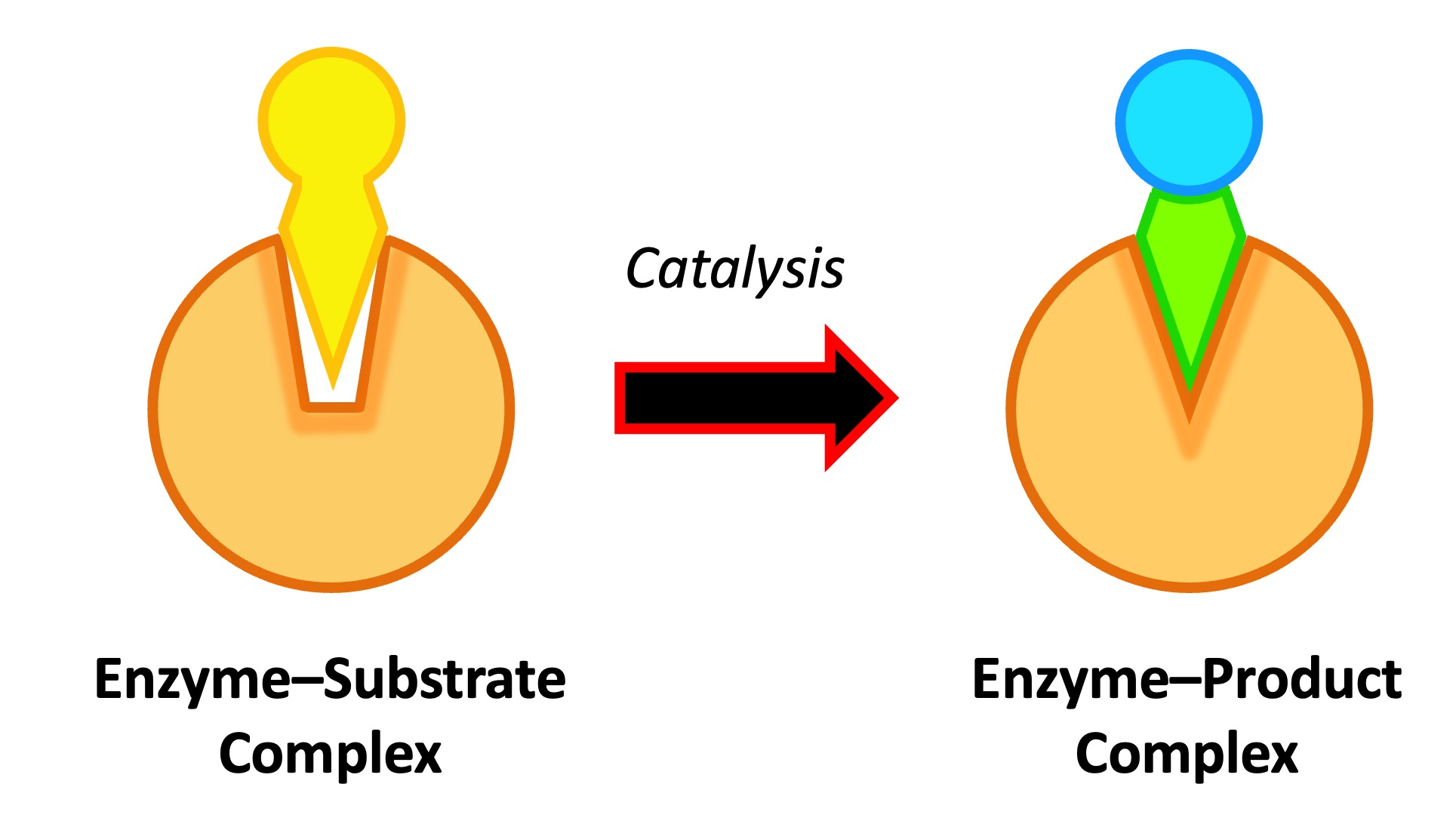
La specificità è un tratto distintivo: ogni enzima di norma riconosce con alta precisione i propri substrati e catalizza una trasformazione definita, incanalando il flusso dei metaboliti lungo itinerari ordinati (Figura 01.13-03). Gli enzimi non vengono consumati: dopo un ciclo catalitico ritornano allo stato iniziale e possono partecipare a ulteriori cicli (Figura 01.13-04). È cruciale sottolineare che la catalisi non modifica la variazione di energia libera \(\Delta G\) né la posizione dell’equilibrio chimico: un enzima accelera allo stesso modo la reazione diretta e quella inversa, consentendo al sistema di raggiungere più rapidamente l’equilibrio senza alterarne il valore.
Dal punto di vista meccanicistico, la riduzione di \(E_a\) può derivare da molteplici contributi che spesso agiscono in sinergia:
- prossimità e orientamento ottimali dei gruppi reattivi, che riducono l’entropia di attivazione;
- stabilizzazione elettrostatica dello stato di transizione mediante residui carichi o dipolari;
- catalisi acido-base generale tramite donazione o accettazione di protoni da parte di residui attivi;
- catalisi covalente con formazione transiente di intermedi legati all’enzima;
- utilizzo di ioni metallici per polarizzare legami o schermare cariche.
Image Gallery

Image Gallery
Image Gallery
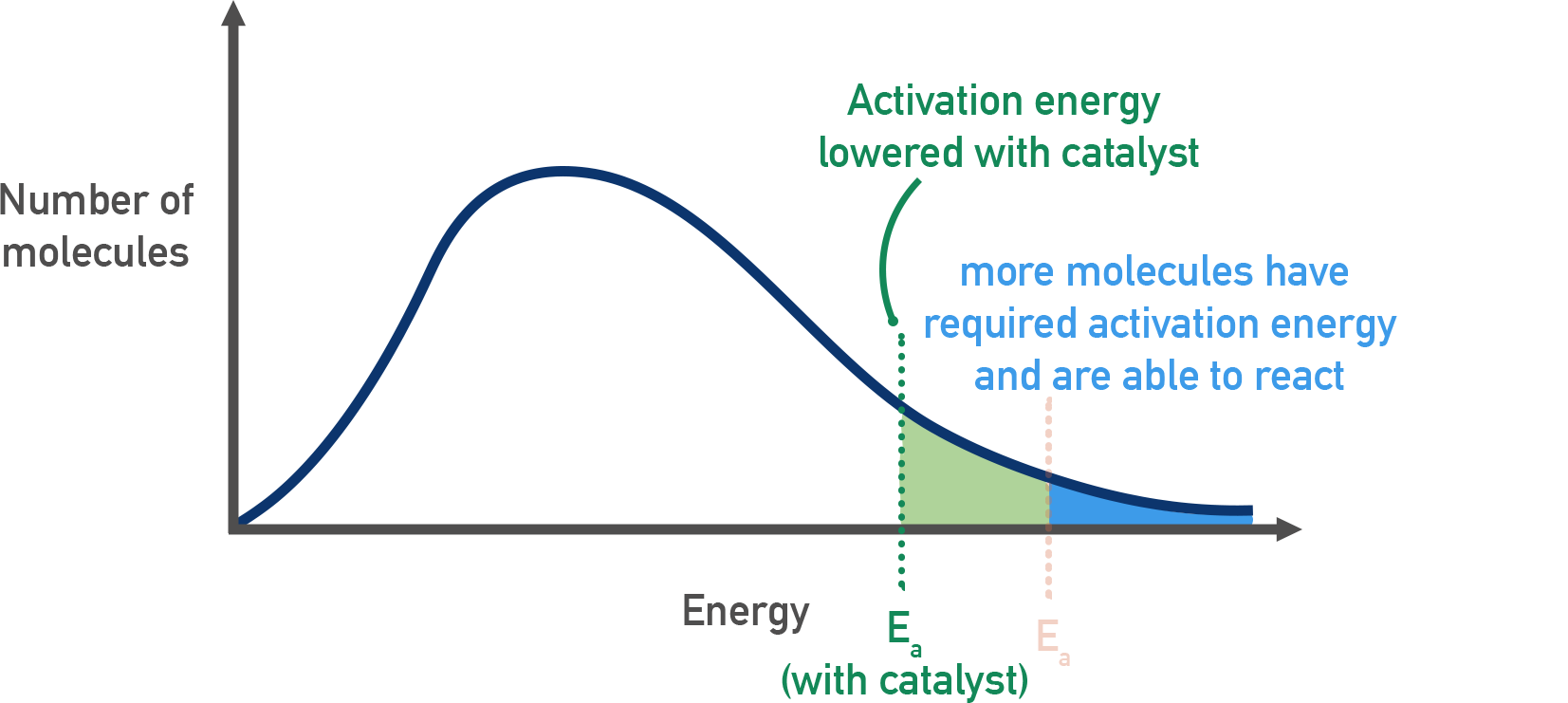
Image Gallery
Image Gallery
Image Gallery
Image Gallery
La seconda legge della termodinamica richiede che, per l’Universo nel suo complesso, il disordine aumenti nel corso di ogni trasformazione (Figura 01.13-13). L’energia disponibile per compiere lavoro chimico è l’energia libera di Gibbs, \(G\). Per le reazioni interessa la variazione \(\Delta G\), che esprime quanto disordine netto viene prodotto quando reagenti si trasformano in prodotti. Per un processo a temperatura costante vale la relazione fondamentale: \[ \Delta G = \Delta H - T\,\Delta S, \] dove \(\Delta H\) è la variazione di entalpia, \(\Delta S\) la variazione di entropia e \(T\) la temperatura assoluta. Una reazione è spontanea se \(\Delta G < 0\), ossia se riduce l’energia libera del sistema (Figura 01.13-05).
Esempi di processi con \(\Delta G\) negativo includono la mescolanza spontanea di due gas inizialmente separati o la decomposizione del perossido di idrogeno, \(2\,\mathrm{H_2O_2} \to 2\,\mathrm{H_2O} + \mathrm{O_2}\), che è termodinamicamente favorita ma, in assenza di catalizzatori come la catalasi o alcuni ioni metallici, procede lentamente. La spontaneità, dunque, non implica velocità: cinetica e termodinamica sono concetti distinti.
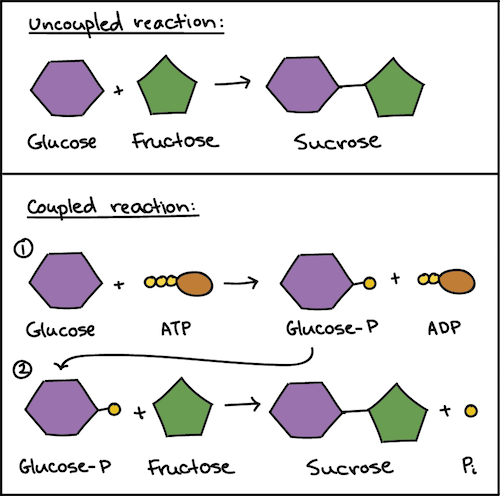
Le reazioni con \(\Delta G > 0\) risultano sfavorite perché aumentano l’ordine del sistema. La formazione di un legame glicosidico tra due monosaccaridi o la polimerizzazione dei nucleotidi in acidi nucleici ne sono esempi: non procedono spontaneamente alle condizioni cellulari. Tali processi possono tuttavia essere resi complessivamente favoriti mediante accoppiamento a reazioni fortemente esoergoniche, per esempio l’idrolisi dell’ATP. Un semplice schema numerico illustra il principio (Figura 01.13-06):
Supponiamo che \(A + B \rightarrow AB\) abbia \(\Delta G^\circ' = +20,0\,\mathrm{kJ\,mol^{-1}}\). Se accoppiamo la reazione all’idrolisi di ATP, con \(\mathrm{ATP} + \mathrm{H_2O} \rightarrow \mathrm{ADP} + \mathrm{P_i}\) e \(\Delta G^\circ' \approx -30,5\,\mathrm{kJ\,mol^{-1}}\), l’insieme risulta: \[ \Delta G^\circ'_\text{complessivo} = +20,0 - 30,5 = -10,5\,\mathrm{kJ\,mol^{-1}}, \] quindi favorito. In pratica, l’accoppiamento avviene tramite la formazione di intermedi “attivati” (per esempio, un intermedio fosforilato) che collegano strettamente i due eventi chimici, spesso all’interno dello stesso sito attivo enzimatico.
Il legame fra termodinamica ed equilibrio è descritto da: \[ \Delta G = \Delta G^\circ' + RT\ln Q,\quad \text{e in equilibrio} \quad \Delta G = 0 \Rightarrow \Delta G^\circ' = -RT\ln K_\mathrm{eq}, \] dove \(Q\) è il quoziente di reazione, \(K_\mathrm{eq}\) la costante di equilibrio, \(R\) la costante dei gas e \(T\) la temperatura. Per esempio, a \(T = 298\,\mathrm{K}\), una reazione con \(\Delta G^\circ' = -5,7\,\mathrm{kJ\,mol^{-1}}\) ha \(K_\mathrm{eq} \approx 10\). Gli enzimi non alterano \(K_\mathrm{eq}\): ne accelerano unicamente il raggiungimento.
La vita è resa possibile dal fatto che gli enzimi, organizzati in vie e complessi multienzimatici, accoppiano in modo efficiente reazioni esoergoniche ed endoergoniche, dirigendo il flusso di materia ed energia verso la costruzione controllata dell’ordine biologico, nel rispetto delle leggi della termodinamica.
Image Gallery
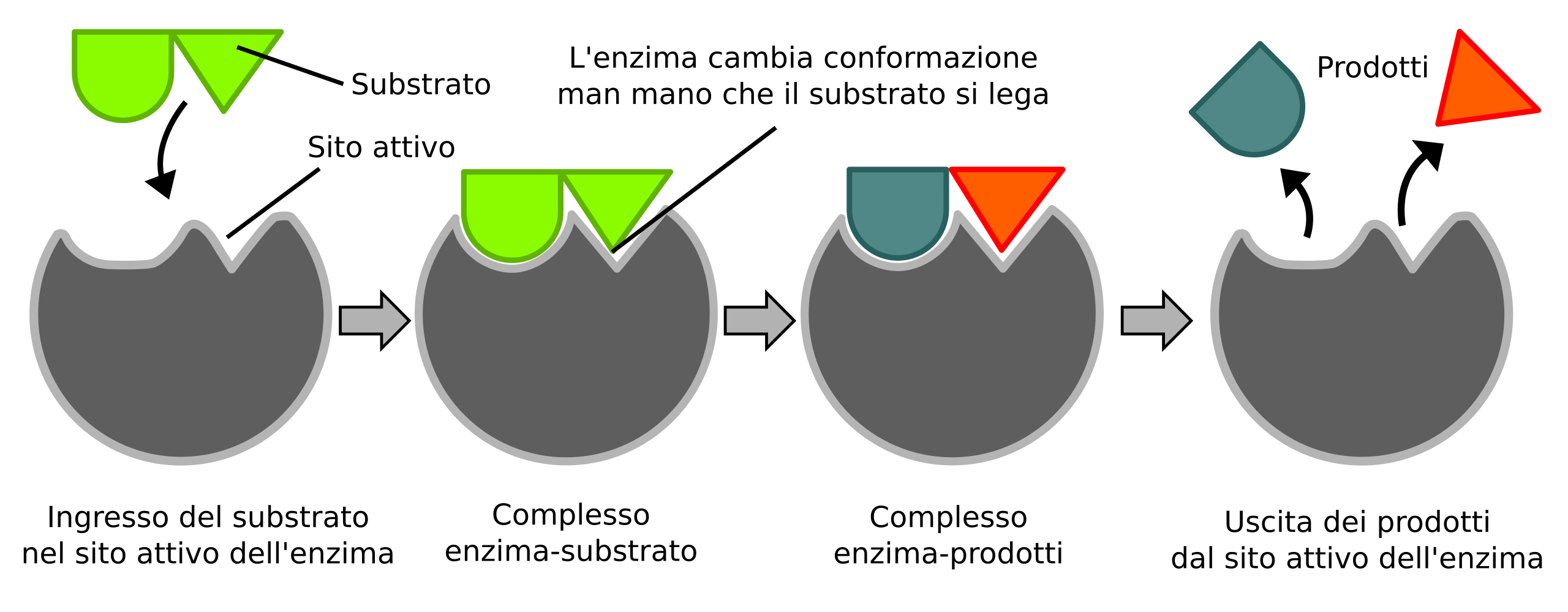
Image Gallery
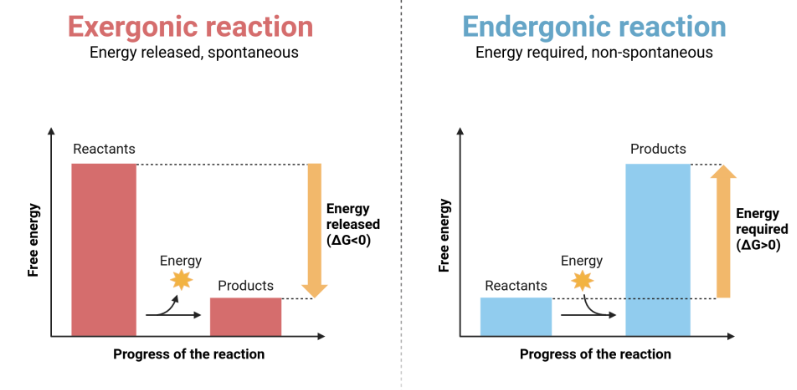
Image Gallery
È immediato capire che un elastico teso, se lasciato libero, si accorcia restituendo calore all’ambiente. Le trasformazioni chimiche sono più articolate: la loro direzione non dipende soltanto dall’energia associata a ciascuna specie, ma anche dalla composizione della miscela di reazione. Un’analogia utile è un’urna con palline di due colori: quando prevalgono le palline blu, l’estrazione casuale di una blu è più probabile che quando le blu sono rare. Analogamente, in una trasformazione favorevole Y → X, mentre la reazione procede, la concentrazione di X aumenta e quella di Y diminuisce; di conseguenza, il rapporto tra substrato e prodotto cambia, e la variazione di energia libera, ΔG, diventa progressivamente meno negativa.
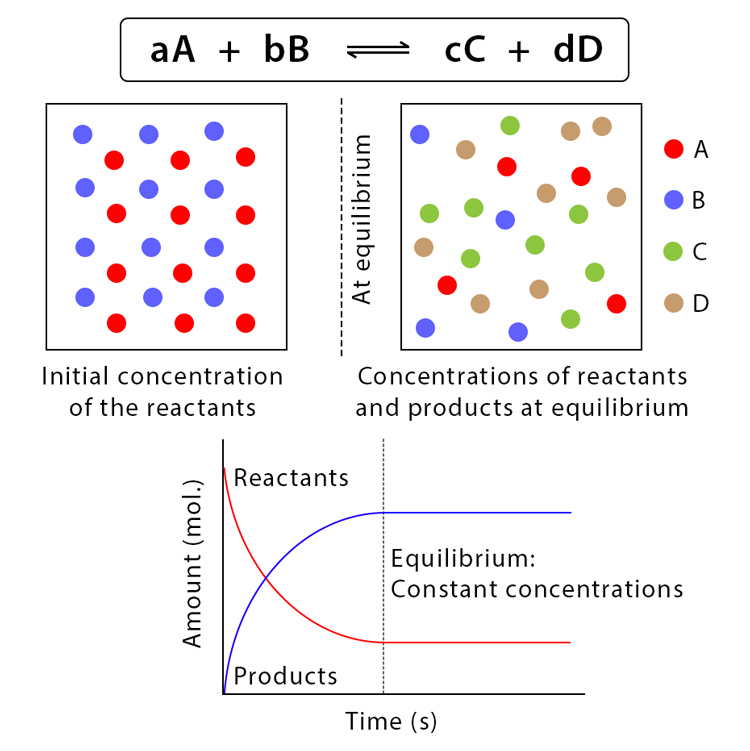
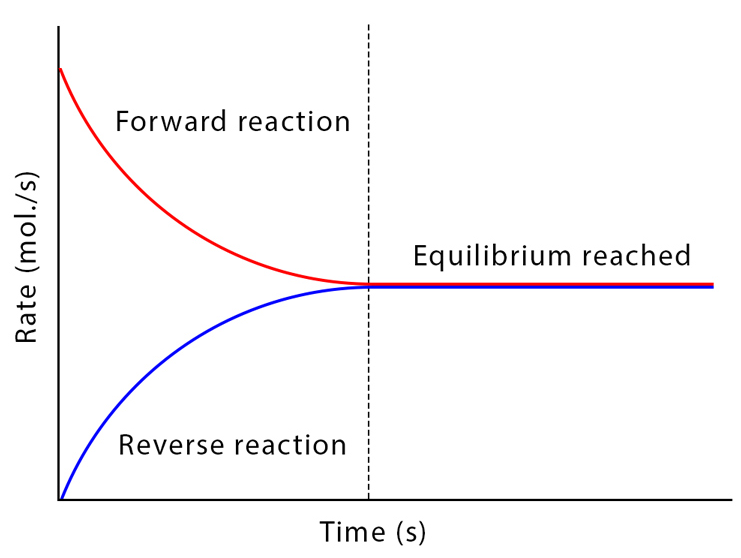
Poiché ΔG dipende dalle concentrazioni istantanee, la reazione rallenta via via che i prodotti si accumulano e i reagenti si consumano, fino a raggiungere uno stato in cui le velocità delle trasformazioni nei due sensi si eguagliano. In questo punto di equilibrio non si osservano variazioni nette delle concentrazioni di reagenti e prodotti e, per definizione, \( \Delta G = 0 \) (Figura 01.13-07). In assenza di lavoro esterno, la reazione non ha tendenza a procedere ulteriormente in alcuna direzione.
Un equilibrio termodinamico completo è incompatibile con i sistemi viventi. Le cellule rimangono lontane dall’equilibrio perché sono sistemi aperti: assorbono nutrienti, eliminano cataboliti e, soprattutto, convogliano i prodotti di una tappa metabolica verso le tappe successive. In tal modo, molte reazioni si mantengono in uno stato stazionario, con flussi continui ma concentrazioni pressoché costanti e \( \Delta G \neq 0 \). In sintesi:
- equilibrio chimico: \( \Delta G = 0 \), velocità diretta e inversa uguali, concentrazioni invarianti;
- stato stazionario metabolico: flussi continui con concentrazioni quasi costanti, ma bilancio di energia libera diverso da zero perché i prodotti sono rimossi o utilizzati in reazioni successive.
Image Gallery
La quantità \( \Delta G \) non è ideale per confrontare l’“energia” di reazioni diverse, perché riflette le concentrazioni correnti dei reagenti. Per valutazioni comparative si adotta la variazione di energia libera standard, \( \Delta G^\circ \), definita nelle condizioni standard in cui tutte le specie hanno attività unitarie (in pratica, concentrazioni pari a 1 mol/L per soluti diluiti) a una temperatura specificata. Così, \( \Delta G^\circ \) dipende solo dalle proprietà intrinseche dei reagenti e non dalla loro concentrazione iniziale. In biochimica è spesso usata la convenzione \( \Delta G^{\circ'} \), standardizzata a pH 7,0.
La relazione generale tra stato reale e standard è:
\[ \Delta G \;=\; \Delta G^\circ \;+\; RT\,\ln Q, \]
dove \( R \) è la costante dei gas, \( T \) la temperatura assoluta e \( Q \) il quoziente di reazione che combina le concentrazioni (o attività) di prodotti e reagenti secondo la stechiometria. Per la trasformazione semplice \( \text{Y} \rightleftharpoons \text{X} \) si ha \( Q = [\text{X}]/[\text{Y}] \) e quindi:
\[ \Delta G \;=\; \Delta G^\circ \;+\; RT\,\ln\!\left(\frac{[\text{X}]}{[\text{Y}]}\right). \]
Quando \( [\text{X}] = [\text{Y}] \) si ha \( Q = 1 \) e, poiché \( \ln 1 = 0 \), segue \( \Delta G = \Delta G^\circ \): in queste condizioni la direzione di marcia dipende esclusivamente dalle proprietà intrinseche della reazione. A 37 °C, \( RT \approx 2,58 \) kJ/mol. Ne consegue, ad esempio, che se \( [\text{X}]/[\text{Y}] = 0{,}01 \) (prodotti scarsi), \( \Delta G = \Delta G^\circ - 11{,}9 \) kJ/mol; la trasformazione verso X risulta quindi più favorevole rispetto alle condizioni standard.
Tutte le reazioni tendono all’equilibrio, caratterizzato da \( \Delta G = 0 \) e \( Q = K \), dove \( K \) è la costante di equilibrio. Inserendo queste condizioni nell’espressione precedente si ottiene la relazione fondamentale:
\[ \Delta G^\circ \;=\; -\,RT\,\ln K. \]
A 37 °C, poiché \( RT = 2{,}58 \) kJ/mol, segue:
\[ \Delta G^\circ \;=\; -\,2{,}58\,\ln K. \]
Convertendo al logaritmo in base 10 si ha:
\[ \Delta G^\circ \;=\; -\,2{,}303\,RT\,\log_{10} K \;=\; -\,5{,}94\,\log_{10} K \quad \text{(kJ/mol, a 37 °C)}. \]
Questa relazione (Tabella 01.13-01) mostra che \( K \) dipende in modo esponenziale da \( \Delta G^\circ \). Ogni variazione di 5,94 kJ/mol in \( \Delta G^\circ \) modifica \( K \) di circa un ordine di grandezza. Esempi numerici:
- se \( K = 100 \), allora \( \Delta G^\circ \approx -11{,}9 \) kJ/mol, con marcata tendenza verso i prodotti;
- se \( K = 0{,}01 \), allora \( \Delta G^\circ \approx +11{,}9 \) kJ/mol, e la reazione standard è sfavorita verso i prodotti;
- se \( K = 1 \), allora \( \Delta G^\circ = 0 \), e alle condizioni standard non esiste preferenza termodinamica per l’uno o l’altro verso.
In pratica, quanto più negativa è \( \Delta G^\circ \), tanto maggiore è l’accumulo del prodotto all’equilibrio.
ΔG° e K
| Costante di equilibrio (K = [X]/[Y]) | Energia libera standard ΔG° (kJ/mol) |
|---|---|
| 10⁵ | –29,7 |
| 10⁴ | –23,8 |
| 10³ | –17,8 |
| 10² | –11,9 |
| 10¹ | –5,9 |
| 1 | 0 |
| 10⁻¹ | +5,9 |
| 10⁻² | +11,9 |
| 10⁻³ | +17,8 |
| 10⁻⁴ | +23,8 |
| 10⁻⁵ | +29,7 |
| Tabella che riporta il legame tra la variazione di energia libera standard (ΔG°) e la costante di equilibrio (K) di una reazione chimica. Quando ΔG° è negativo, il processo è favorito e procede spontaneamente verso la formazione dei prodotti; al contrario, valori positivi di ΔG° indicano una reazione sfavorita. I dati riportati di seguito si riferiscono a condizioni standard a 37 °C. | |
Nelle reti cellulari, molte trasformazioni coinvolgono più specie. Per la reazione bimolecolare \( \text{A} + \text{B} \rightleftharpoons \text{AB} \), la costante di equilibrio è:
\[ K \;=\; \frac{[\text{AB}]}{[\text{A}]\,[\text{B}]}. \]
Le concentrazioni dei reagenti compaiono moltiplicate perché la formazione di AB richiede l’urto tra A e B; la frequenza di collisione è proporzionale al prodotto \( [\text{A}]\,[\text{B}] \) (Figura 01.13-08). La relazione tra \( K \) e \( \Delta G^\circ \) rimane invariata: a 37 °C, \( \Delta G^\circ = -5{,}94\,\log_{10} K \) (Tabella 01.13-01).
La formulazione si estende a qualsiasi stechiometria. Per una reazione generale:
\[ \nu_A\,\text{A} + \nu_B\,\text{B} \rightleftharpoons \nu_C\,\text{C} + \nu_D\,\text{D}, \]
si definiscono il quoziente di reazione e la costante di equilibrio come:
\[ Q \;=\; \frac{[\text{C}]^{\nu_C}[\text{D}]^{\nu_D}}{[\text{A}]^{\nu_A}[\text{B}]^{\nu_B}}, \qquad K \;=\; \frac{[\text{C}]^{\nu_C}[\text{D}]^{\nu_D}}{[\text{A}]^{\nu_A}[\text{B}]^{\nu_B}}, \]
e vale sempre \( \Delta G = \Delta G^\circ + RT\,\ln Q \) e, all’equilibrio, \( \Delta G^\circ = -RT\,\ln K \). Un esempio: per \( 2\text{A} + \text{B} \rightleftharpoons \text{A}_2\text{B} \) con \( K = 5{,}0 \times 10^2 \), a 37 °C si ottiene \( \Delta G^\circ \approx -15{,}3 \) kJ/mol; all’equilibrio, questo implica \( [\text{A}_2\text{B}] \approx 500\, [\text{A}]^2[\text{B}] \), quindi la reazione è fortemente spostata verso il complesso.
In sistemi biologici reali, K e \( \Delta G^\circ \) sono determinati da proprietà molecolari fondamentali (energie di legame, interazioni solvente–solute, ecc.). Le cellule modulano la direzione effettiva delle trasformazioni regolando \( Q \) tramite il controllo delle concentrazioni e il collegamento di reazioni in sequenza, in modo che i prodotti di una tappa siano rapidamente rimossi come substrati della successiva, mantenendo il flusso lontano dall’equilibrio.
Image Gallery
ΔG° e K
| Costante di equilibrio (K = [X]/[Y]) | Energia libera standard ΔG° (kJ/mol) |
|---|---|
| 10⁵ | –29,7 |
| 10⁴ | –23,8 |
| 10³ | –17,8 |
| 10² | –11,9 |
| 10¹ | –5,9 |
| 1 | 0 |
| 10⁻¹ | +5,9 |
| 10⁻² | +11,9 |
| 10⁻³ | +17,8 |
| 10⁻⁴ | +23,8 |
| 10⁻⁵ | +29,7 |
| Tabella che riporta il legame tra la variazione di energia libera standard (ΔG°) e la costante di equilibrio (K) di una reazione chimica. Quando ΔG° è negativo, il processo è favorito e procede spontaneamente verso la formazione dei prodotti; al contrario, valori positivi di ΔG° indicano una reazione sfavorita. I dati riportati di seguito si riferiscono a condizioni standard a 37 °C. | |
La variazione di energia libera governa non solo le trasformazioni che implicano la rottura e la formazione di legami covalenti, ma anche i fenomeni di riconoscimento e associazione tra molecole mediati da interazioni non covalenti. Due partner molecolari tendono a formare un complesso stabile quando il contributo di energia libera dell’associazione risulta negativo: in tal caso, lo stato legato possiede energia libera inferiore rispetto alla somma degli stati separati. L’efficacia di tali interazioni è cruciale per i processi cellulari, tra cui:
- il legame tra enzimi e substrati, alla base della catalisi biochimica;
- il riconoscimento tra fattori di trascrizione e DNA nei circuiti di regolazione genica;
- l’assemblaggio di complessi multiproteici, sia con funzione strutturale sia con funzione catalitica o regolatoria.
La costante di equilibrio \(K\) che descrive una reazione chimica si applica in modo del tutto analogo all’associazione non covalente A + B ⇌ AB. All’equilibrio, quando la velocità di associazione eguaglia la velocità di dissociazione, le concentrazioni \([A]\), \([B]\) e \([AB]\) consentono di calcolare \(K\) secondo:
\[ K \;=\; \frac{[AB]}{[A][B]} \qquad \text{(Figura 01.13-08)} \]
L’entità di \(K\) riflette la forza e la specificità del legame: valori maggiori di \(K\) denotano maggiore stabilità del complesso e più marcata differenza di energia libera tra stato associato e dissociato. Per una coppia data, l’energia libera standard di legame è:
\[ \Delta G^\circ_{\text{leg}} \;=\; -\,RT \ln K \,, \]
per cui un aumento dell’energia di legame (più interazioni favorevoli) produce un incremento esponenziale di \(K\). Anche un piccolo numero di interazioni deboli addizionali può determinare variazioni sostanziali di affinità: come illustrato nella (Figura 01.13-09), l’eliminazione di alcuni legami a idrogeno, equivalente a una perdita di circa 12,0 kJ/mol di energia di legame, può ridurre il complesso all’equilibrio di oltre due ordini di grandezza, dato che \(\Delta\Delta G^\circ \approx 12,0\) kJ/mol corrisponde a \(\exp(\Delta\Delta G^\circ/RT) \approx 10^2\)–\(10^3\) a 298 K.
Per completezza, si usa spesso anche la costante di dissociazione \(K_d = 1/K\), direttamente interpretabile come concentrazione alla quale metà dei siti di legame risultano occupati; più piccolo è \(K_d\), maggiore è l’affinità. Parametri fisico‑chimici come forza ionica, pH e temperatura modulano \(K\) e \(K_d\) influenzando l’equilibrio tra contributi entalpici (es. legami a idrogeno, interazioni elettrostatiche, di van der Waals) ed entropici (effetti di rilascio dell’acqua, costrizione conformazionale).
Image Gallery
Image Gallery

Un tema centrale dell’organizzazione metabolica è come la cellula realizzi trasformazioni sfavorite dal punto di vista energetico. Una strategia generale consiste nell’accoppiare reazioni sfavorite a reazioni fortemente favorite, in modo che la somma delle variazioni di energia libera risulti negativa. Se consideriamo due reazioni in sequenza, X ⇌ Y e Y ⇌ Z, la variazione complessiva standard di energia libera è additiva:
\[ \Delta G^\circ_{\text{tot}} \;=\; \Delta G^\circ_{X \to Y} \;+\; \Delta G^\circ_{Y \to Z} \,. \]
Ad esempio, se \(\Delta G^\circ_{X \to Y} = +18\) kJ/mol e \(\Delta G^\circ_{Y \to Z} = -45\) kJ/mol, allora \(\Delta G^\circ_{X \to Z} = -27\) kJ/mol: la sequenza complessiva è termodinamicamente favorita. Poiché 1 mole corrisponde a circa \(6 \cdot 10^{23}\) molecole, questi bilanci energetici macroscopici riflettono l’andamento medio di un enorme numero di eventi molecolari.
Operativamente, l’enzima che catalizza Y ⇌ Z “prosciuga” l’intermedio Y non appena si forma, fungendo da sifone cinetico che trascina la conversione di X a Z (Figura 01.13-10). Molte tappe di vie cataboliche complesse, come l’ossidazione degli zuccheri fino a CO₂ e H₂O, includono passaggi sfavoriti che procedono rapidamente perché la somma di tutte le \(\Delta G^\circ\) risulta fortemente negativa. In altri contesti, l’obiettivo può essere arrestare il percorso al livello di Y. In tal caso, l’accoppiamento energetico si ottiene frequentemente tramite la formazione e l’impiego di intermedi ad alta energia o di molecole-trasportatrici attivate (per esempio ATP, NAD(P)H, acetil‑CoA), che trasferiscono selettivamente gruppi chimici o equivalenti riducenti tra reazioni distinte.
Image Gallery
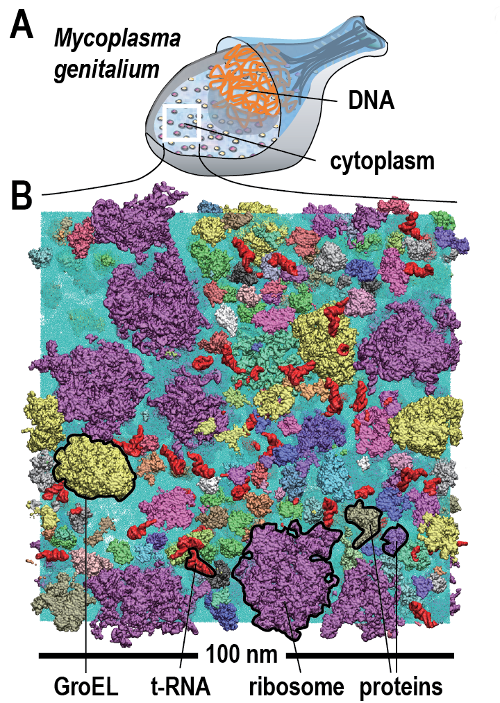
Nel citosol, un milieu densamente occupato da macromolecole di forme e dimensioni diverse (Figura 01.13-11), enzimi e substrati devono incontrarsi nonostante le concentrazioni spesso modeste. Il motore di tali incontri è il moto browniano: l’agitazione termica determina traiettorie di tipo cammino aleatorio, con collisioni incessanti fra specie molecolari (Figura 01.13-12). Per una molecola piccola, i coefficienti di diffusione intracellulari sono tipicamente dell’ordine di 100–300 μm²/s, solo poco inferiori all’acqua; il tempo medio per percorrere 10 μm può essere stimato dall’equazione della diffusione \(\langle r^2 \rangle = 6Dt\), che fornisce \(t \approx L^2/(6D)\). Con \(L=10\) μm e \(D=200\) μm²/s, si ottiene \(t \approx 0,08\)–0,2 s, a conferma dell’efficienza della diffusione su distanze cellulari.
Le proteine, più voluminose, diffondono più lentamente; di conseguenza, la frequenza di incontro tra un enzima e il suo substrato dipende fortemente dalla concentrazione del substrato. Per concentrazioni intracellulari dell’ordine di 0,3 mM, a fronte di una concentrazione dell’acqua di circa 55,5 M, si ha all’incirca una molecola di substrato ogni ~2·10⁵ molecole di H₂O; ciononostante, un singolo sito attivo può essere colpito casualmente da \(10^5\)–\(10^6\) molecole di substrato al secondo. Per una concentrazione dieci volte inferiore (0,03 mM), il numero di collisioni resta dell’ordine di \(10^4\)–\(10^5\) s⁻¹. Questi tassi elevati sostengono velocità catalitiche tipiche dell’ordine di \(10^3\) s⁻¹ per molti enzimi.
Esiste inoltre un limite superiore fissato dalla diffusione per l’associazione bimolecolare, descritto dal modello di Smoluchowski: i valori massimi di \(k_{\text{on}}\) si collocano intorno a \(10^8\)–\(10^9\) M⁻¹ s⁻¹ in soluzione acquosa. Al di sotto di tale limite, la chimica del sito attivo e la corretta orientazione delle molecole incidenti determinano l’efficienza con cui le collisioni diventano complessi produttivi.
Image Gallery
Image Gallery
Ogni reazione catalizzata inizia con la formazione del complesso enzima–substrato. La permanenza del substrato nel sito attivo deve essere sufficientemente lunga da consentire il superamento della barriera di attivazione e la trasformazione chimica. La stabilizzazione deriva dalla somma di molte interazioni deboli (legami a idrogeno, interazioni elettrostatiche schermate, contatti idrofobici e di van der Waals), ciascuna di modesta entità ma cooperanti. Se le superfici in contatto sono poco complementari, si instaurano pochi contatti favorevoli, la stabilizzazione non supera l’energia termica e la dissociazione è rapida. Come suggerito in (Figura 01.13-09), anche una modesta variazione nel numero o nella geometria di tali legami può alterare drasticamente la probabilità di formazione del complesso.
Questa sensibilità è vantaggiosa: impedisce associazioni spurie con substrati non corretti e favorisce invece i partner “cognati”, per i quali l’adattamento geometrico e chimico ottimizza il numero di interazioni deboli. Spesso, l’enzima contribuisce tramite aggiustamenti conformazionali indotti dal legame (induced fit), che allineano i gruppi catalitici e il substrato nello stato di transizione. Pur riducendo l’energia di attivazione, l’enzima non altera l’equilibrio termodinamico della reazione: la diminuzione della barriera riguarda allo stesso modo il processo in avanti e quello inverso, cosicché la posizione dell’equilibrio (e dunque \(\Delta G^\circ\)) resta invariata (Figura 01.13-01) e (Figura 01.13-13). In sintesi, gli enzimi aumentano la velocità con cui l’equilibrio è raggiunto senza modificarne il valore.
Image Gallery
Image Gallery
Image Gallery


