


Gli enzimi accelerano le reazioni metaboliche abbassandone la barriera energetica
TOPICS
Definizione

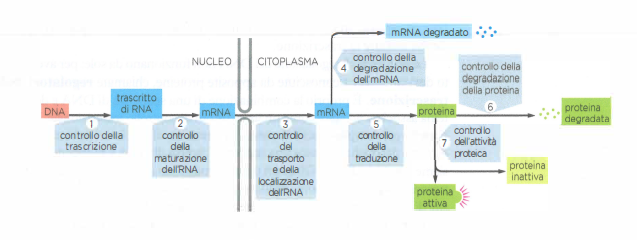
I regolatori della trascrizione stabiliscono se un gene venga attivato o silenziato, determinando l’avvio della sintesi dell’RNA. Nella maggior parte degli organismi, questo livello di controllo è ampiamente diffuso. Tuttavia, lungo il percorso che conduce dal DNA alla proteina esistono ulteriori snodi decisionali che intervengono dopo l’inizio della trascrizione, offrendo alle cellule ulteriori gradi di libertà per modulare quantità e attività dei prodotti genici (Figura 03.12-06). Questi meccanismi post-trascrizionali contribuiscono in modo sostanziale alla regolazione dell’espressione della maggior parte dei geni. Tra essi rientrano processi già menzionati, come lo splicing alternativo, capace di generare varianti proteiche distinte a partire da un unico locus, e le modificazioni post-traduzionali, che influenzano stabilità, localizzazione e funzione delle proteine mature. Altri passaggi, di crescente rilievo, consentono di rimodellare l’espressione genica una volta che la trascrizione è iniziata:
- maturazione del trascritto primario (capping, splicing, poliadenilazione);
- editing dell’RNA e sorveglianza della qualità, come la degradazione mediata da codoni di stop prematuri (NMD);
- esportazione nucleare e localizzazione subcellulare degli mRNA;
- controllo dell’efficienza di traduzione e del reclutamento ribosomiale;
- determinazione della stabilità dell’mRNA tramite deadenilazione, decapping e vie di decadimento;
- regolazione post-traduzionale delle proteine, che ne modula concentrazione e attività.
Image Gallery
La stabilità di un mRNA, spesso espressa come emivita \(t_{1/2}\), e la sua efficienza di traduzione dipendono da elementi in cis localizzati nelle regioni non tradotte (UTR) a 5′ e a 3′. Queste sequenze fungono da piattaforme di riconoscimento per proteine leganti l’RNA o per piccoli RNA regolatori, e veicolano istruzioni su se, quando e con quale frequenza un trascritto venga convertito in proteina.
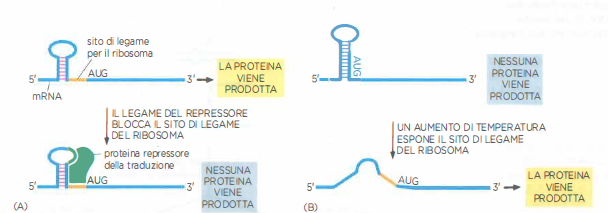
Nei batteri, una breve sequenza a monte del codone di inizio AUG, la sede di legame ribosomiale, si appaia con l’RNA 16S della piccola subunità del ribosoma, posizionando correttamente l’AUG nel sito di inizio. Poiché tale interazione è necessaria per un avvio accurato, essa costituisce un punto privilegiato di controllo: la cellula può inibire o favorire la traduzione mascherando o esponendo la regione di legame ribosomiale mediante strutture secondarie dell’RNA, proteine repressorie o piccoli RNA regolatori (Figura 03.12-01). In diversi casi, interruttori conformazionali dell’RNA, come riboswitch o termometri RNA, modulano l’accessibilità del sito in risposta a metaboliti o temperatura.
Negli eucarioti, l’inizio della traduzione avviene tipicamente tramite scansione a partire dal cappuccio 5′ fino al primo AUG in un contesto favorevole (sequenza di Kozak). Elementi nella 5′ UTR, incluse strutture secondarie, piccoli open reading frame a monte (uORF) e motivi specifici, possono reclutare proteine repressorie che impediscono al ribosoma di riconoscere l’AUG iniziale. Quando cambiano le condizioni cellulari, la rimozione o inattivazione di tali repressori consente il reclutamento ribosomiale e l’avvio della traduzione. Questi controlli in cis interagiscono con regolazioni globali dei fattori di inizio, creando una rete multilivello di modulazione traducente.
Image Gallery
Gli RNA cellulari non si limitano ai trascritti codificanti proteine. Oltre a tRNA e rRNA, dotati di funzioni strutturali e catalitiche indispensabili per la sintesi proteica, e all’RNA della telomerasi, essenziale per la replicazione completa dei cromosomi eucariotici, molti organismi, in particolare piante e animali, esprimono migliaia di RNA non codificanti con ruoli regolatori. Questi RNA di regolazione costituiscono una classe eterogenea che agisce su più livelli dell’espressione genica:
- microRNA (miRNA), piccoli regolatori endogeni che riconoscono mRNA complementari per modulare stabilità e traduzione;
- piccoli RNA interferenti (siRNA), spesso coinvolti nella difesa contro elementi mobili e RNA esogeni, con appaiamento pressoché perfetto ai bersagli;
- lunghi RNA non codificanti (lncRNA), che fungono da guide, impalcature o decoy per complessi proteici, anche in relazione alla regolazione cromatinica.
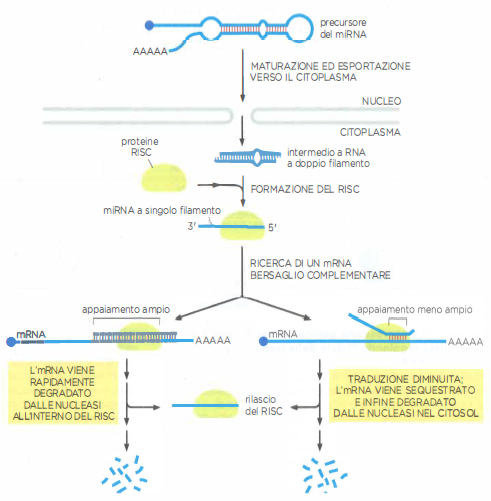
I microRNA (miRNA) sono piccoli RNA regolatori, lunghi circa 22 nucleotidi, che modulano l’espressione genica riconoscendo tramite appaiamento di basi specifiche sequenze in mRNA bersaglio, con effetti sulla loro stabilità e sulla frequenza di traduzione. La biogenesi dei miRNA inizia con la trascrizione di precursori primari (pri-miRNA) da parte dell’RNA polimerasi II; questi trascritti, spesso cappati e poliadenilati, vengono processati nel nucleo dal complesso Drosha–DGCR8 per generare capelli pre-miRNA. Dopo l’esportazione nel citoplasma mediata da Exportin‑5, l’enzima Dicer rifinisce i pre-miRNA in duplex di ~22 nucleotidi. Uno dei due filamenti (filamento guida) viene poi incorporato, insieme a una proteina Argonaute, nel complesso RISC (RNA-Induced Silencing Complex), che esplora il citoplasma alla ricerca di mRNA con sequenze complementari (Figura 03.12-02).
Il riconoscimento del bersaglio è guidato in larga misura dalla regione “seed” del miRNA (posizioni 2–8). Quando l’appaiamento è esteso, il complesso contenente Argonaute può tagliare l’mRNA; quando l’appaiamento è parziale, prevale la repressione traducente seguita da rimozione della coda poli(A) e del cappuccio 5′, eventi che conducono alla degradazione del trascritto. Spesso gli mRNA repressi vengono convogliati in domini citoplasmatici deputati alla loro demolizione. Dopo il rilascio del bersaglio, RISC è riciclabile e può agire in serie su altri mRNA, rendendo il silenziamento altamente efficiente.
Nel genoma umano sono codificati diverse centinaia di miRNA, e ciascuno può regolare numerosi trascritti; nel loro complesso essi influenzano una quota sostanziale dei geni codificanti proteine, con stime che raggiungono circa un terzo del totale. L’impatto dei miRNA si manifesta in processi quali differenziamento, risposta allo stress e omeostasi tissutale; di converso, alterazioni della loro espressione o funzione si associano a numerose condizioni patologiche. In sintesi, il circuito miRNA–RISC costituisce un asse centrale del controllo post-trascrizionale, capace di integrare segnali cellulari e rimodulare in modo rapido e preciso il profilo proteico della cellula.
Image Gallery
Una parte dell’apparato che genera e gestisce i miRNA partecipa anche a un sistema di sorveglianza molecolare altamente efficiente. Questo assetto utilizza il riconoscimento di RNA a doppio filamento (dsRNA), struttura rara nei trascritti cellulari ordinari ma comune nei cicli replicativi dei virus e nel movimento di elementi trasponibili, per attivare meccanismi di silenziamento mirato. Gli RNA estranei vengono così neutralizzati tramite l’interferenza a RNA (RNAi), prevenendo la propagazione di sequenze potenzialmente dannose.
Nella fase iniziale, lunghi dsRNA vengono frammentati da Dicer in piccoli duplex di circa 21–23 nucleotidi, con tipici sbordi 3' di 2 nucleotidi (Figura 03.12-02). I prodotti di Dicer, i piccoli RNA interferenti (siRNA, small interfering RNA), vengono poi caricati in complessi RISC (RNA-Induced Silencing Complex), che condividono componenti con la via dei miRNA, tra cui proteine Argonaute. Durante l’attivazione del RISC, uno dei due filamenti del siRNA (passenger strand) viene rimosso, mentre il filamento guida orienta il complesso verso RNA complementari, che vengono poi clivati dall’endomuclease associata a Argonaute o degradati da ribonucleasi reclutate (Figura 03.12-03). In tal modo, l’RNA virale o trasponibile complementare viene rapidamente eliminato dallo spazio citoplasmatico.
Oltre al silenziamento post-trascrizionale, l’RNAi può operare a livello trascrizionale. In diversi eucarioti, i siRNA generati da Dicer vengono assemblati in complessi RITS (RNA-Induced Transcriptional Silencing). Guidato dal proprio siRNA a singolo filamento, il RITS riconosce sequenze complementari su trascritti nascenti associati a RNA polimerasi attive e ingaggia fattori enzimatici che modificano gli istoni vicini, promuovendo marchi di eterocromatina (ad esempio metilazioni su H3K9) e il compattamento della cromatina (Figura 03.12-04). La formazione di eterocromatina che ne deriva ostacola l’inizio e l’allungamento della trascrizione, limitando la mobilità degli elementi trasponibili e la produzione di RNA estranei.
La risposta mediata da RNAi è ampiamente distribuita in natura e mostra una chiara antichità evolutiva: è documentata in lieviti, piante, nematodi e insetti, tra gli altri. In alcuni taxa, la via viene potenziata da RNA polimerasi RNA-dipendenti, che amplificano i siRNA a partire da trascritti target, e può propagarsi a distanza tra tessuti. Nelle piante, per esempio, segnali di RNAi diffondono attraverso plasmodesmi e floema, consentendo una protezione sistemica anche quando l’infezione è ancora localizzata. Un’analogia funzionale, seppur di natura molecolare differente, può essere tracciata con l’immunità adattativa dei vertebrati: in entrambi i casi si generano molecole a elevata specificità (siRNA o anticorpi) capaci di riconoscere e neutralizzare un bersaglio determinato, riducendo il danno all’ospite:
- Riconoscimento del dsRNA come segnale di pericolo, frequente in virus ed elementi mobili;
- Processamento da parte di Dicer in siRNA di 21–23 nucleotidi con sbordi 3' di 2 nucleotidi (Figura 03.12-02);
- Caricamento nel RISC, eliminazione del filamento non guida e clivaggio dell’RNA complementare tramite Argonaute (Figura 03.12-03);
- Silenziamento trascrizionale mediato da RITS e instaurazione di eterocromatina nelle regioni bersaglio (Figura 03.12-04);
- Contenimento di virus ed elementi trasponibili, con possibilità di amplificazione e diffusione del segnale in alcune specie.
Esempio applicativo: in una pianta perenne esposta a un virus a RNA, l’enzima Dicer-like processa i dsRNA virali in siRNA, che vengono amplificati da un’RNA polimerasi RNA-dipendente. I siRNA si muovono verso foglie non ancora infette, dove, una volta caricati nei RISC locali, degradano trascritti virali appena sintetizzati, contribuendo a una resistenza generalizzata dell’organismo.
È utile distinguere il comportamento dei siRNA da quello dei miRNA: i primi tendono a riconoscere il bersaglio con complementarietà pressoché completa e promuovono il clivaggio endonucleolitico, mentre i miRNA spesso tollerano disappaiamenti e, in tal caso, reprimono prevalentemente la traduzione o indirizzano a decadenza il trascritto con meccanismi diversi. In molte linee germinali animali, un’ulteriore classe di piccoli RNA, i piRNA, coopera con proteine PIWI per reprimere trasposoni, sottolineando la diversificazione evolutiva delle vie di difesa basate su RNA.
Image Gallery
Image Gallery
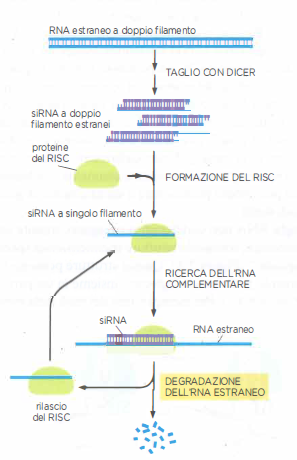
Image Gallery
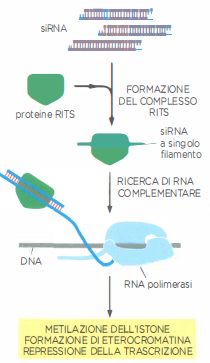
Image Gallery
Image Gallery
Image Gallery
All’estremo opposto per dimensioni rispetto ai piccoli RNA si collocano i lunghi RNA non codificanti (lncRNA), convenzionalmente definiti come trascritti superiori a 200 nucleotidi. Nel genoma dei mammiferi sono presenti migliaia di loci che generano lncRNA, molti dei quali mostrano pattern di espressione specifici per cellula, tessuto e stadio di sviluppo. Sebbene le funzioni siano eterogenee e in molti casi ancora da chiarire, è emerso che i lncRNA possono agire come guide, impalcature, decoy e modulatori dell’architettura cromatinica.
Un paradigma funzionale è offerto da Xist, un lncRNA di circa 17 000 nucleotidi che governa l’inattivazione del cromosoma X nelle cellule dei mammiferi femmina. In una finestra precoce dello sviluppo, Xist viene trascritto da uno solo dei due cromosomi X e rimane associato in cis al cromosoma d’origine. Il rivestimento del cromosoma da parte di Xist favorisce il reclutamento di complessi di rimodellamento della cromatina e di enzimi modificatori degli istoni, con l’accumulo di segni repressivi e la progressiva condensazione in eterocromatina stabile, l’analogo del corpo di Barr (Figura 03.12-03). Questo assetto garantisce un silenziamento durevole dei geni sul cromosoma inattivo, preservando il bilanciamento del dosaggio genico tra i sessi.
Molti lncRNA esercitano funzioni tramite la loro struttura secondaria e terziaria, che consente interazioni multivalenti con proteine e acidi nucleici. In tale veste, possono fungere da piattaforme per assemblare complessi enzimatici o per spazializzare reazioni in distretti subnucleari o citoplasmatici (Figura 03.12-05). Un esempio emblematico è l’RNA della telomerasi, che contribuisce sia come stampo per l’estensione delle estremità cromosomiche sia come impalcatura per l’organizzazione delle subunità proteiche della holoenzima, coordinandone attività e stabilità:
- Regolazione cis e trans dell’espressione genica, guidando complessi di rimodellamento verso loci specifici;
- Funzione di scaffolding, riunendo più proteine in complessi funzionali localizzati (Figura 03.12-05);
- Ruolo di decoy, sequestrando fattori regolatori e modulandone la disponibilità;
- Contributo all’organizzazione nucleare, inclusa la formazione di domini e corpi subnucleari;
- Supporto a processi enzimatici, come nel caso dell’RNA della telomerasi, coordinando subunità e attività catalitiche.
Alcuni lncRNA mostrano una forte specificità temporale e topologica: possono essere prodotti in risposta a segnali di differenziamento o stress e restare confinati vicino al sito di trascrizione, dove influenzano la cromatina nei dintorni, oppure agire a distanza su cromosomi diversi. La loro azione si integra con reti proteiche e con altre classi di RNA regolatori, suggerendo che il genoma eucariotico non sia soltanto un archivio di codici per proteine, ma anche una matrice di istruzioni distribuite che orchestrano quando, dove e in quale misura le informazioni vengano espresse.
Nel loro insieme, i lncRNA ampliano il repertorio delle strategie con cui le cellule regolano la funzione genica e l’assetto strutturale del genoma. Che agiscano guidando modificazioni covalenti degli istoni, stabilizzando complessi multiproteici o modulando l’accessibilità della cromatina, essi contribuiscono a definire i programmi di crescita, differenziamento e omeostasi dei mammiferi.
Image Gallery
Image Gallery
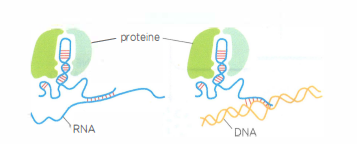
Image Gallery


