Durante la fosforilazione ossidativa, il processo della chemiosmosi accoppia il trasporto di elettroni alla sintesi dell’ATP
TOPICS
Definizione

Il genoma umano custodisce un patrimonio informativo vastissimo sulla nostra identità biologica e sulla storia evolutiva della specie (Figura 04.05-01). Le sue \(3{,}2 \times 10^9\) coppie di nucleotidi, organizzate in 23 insiemi cromosomici (22 autosomi e una coppia sessuale, X e Y), contengono le istruzioni necessarie alla costruzione e al funzionamento dell’organismo umano. Fino a pochi decenni fa era legittimo domandarsi se ricavare l’intera sequenza del DNA umano — l’elenco completo dei nucleotidi — fosse davvero imprescindibile. L’impresa, complessa e costosa, è stata affrontata da un consorzio internazionale che, nell’arco di quasi dieci anni e con un investimento di circa 3 miliardi di dollari, ha prodotto la prima mappa completa di riferimento del nostro patrimonio genetico. Il ritorno scientifico è stato enorme: la disponibilità di una sequenza di riferimento ha trasformato ogni area della biologia e della medicina, consentendo di formulare e verificare ipotesi sulle funzioni del genoma e sulla sua evoluzione su basi quantitative e riproducibili. L’avvio del Progetto Genoma Umano nei primi anni ’90 ha innescato progressi tecnologici continui. Oggi, grazie a piattaforme di sequenziamento ad alta processività e a infrastrutture di calcolo capaci di gestire dati su scala petabyte, è possibile ottenere la sequenza completa di un genoma umano in pochi giorni con un costo di circa 1000 dollari. Il flusso mondiale di dati genomici — generato in progetti che confrontano migliaia di genomi — consente di studiare con precisione senza precedenti la variazione genetica, le basi molecolari delle malattie e le traiettorie evolutive umane. Dopo le prime bozze pubblicate nel 2001 e la versione di riferimento del 2004, ulteriori traguardi hanno colmato molte lacune residue, fino a completare interi cromosomi in modo contiguo. Parallelamente, la costruzione di riferimenti “pangenomici” ha iniziato a rappresentare in modo più fedele la diversità strutturale presente nelle popolazioni umane. La quantità e la qualità delle informazioni oggi disponibili sono tali da offrire risposte a questioni antiche — che cosa ci accomuna come specie e che cosa ci differenzia come individui — e, insieme, da aprire nuovi interrogativi. Mentre l’analisi integrata di queste risorse procede, risultati recenti hanno già ridisegnato in profondità la nostra comprensione dell’architettura genomica, della regolazione dell’espressione genica e dei meccanismi alla base dell’adattamento e della patologia.
Image Gallery
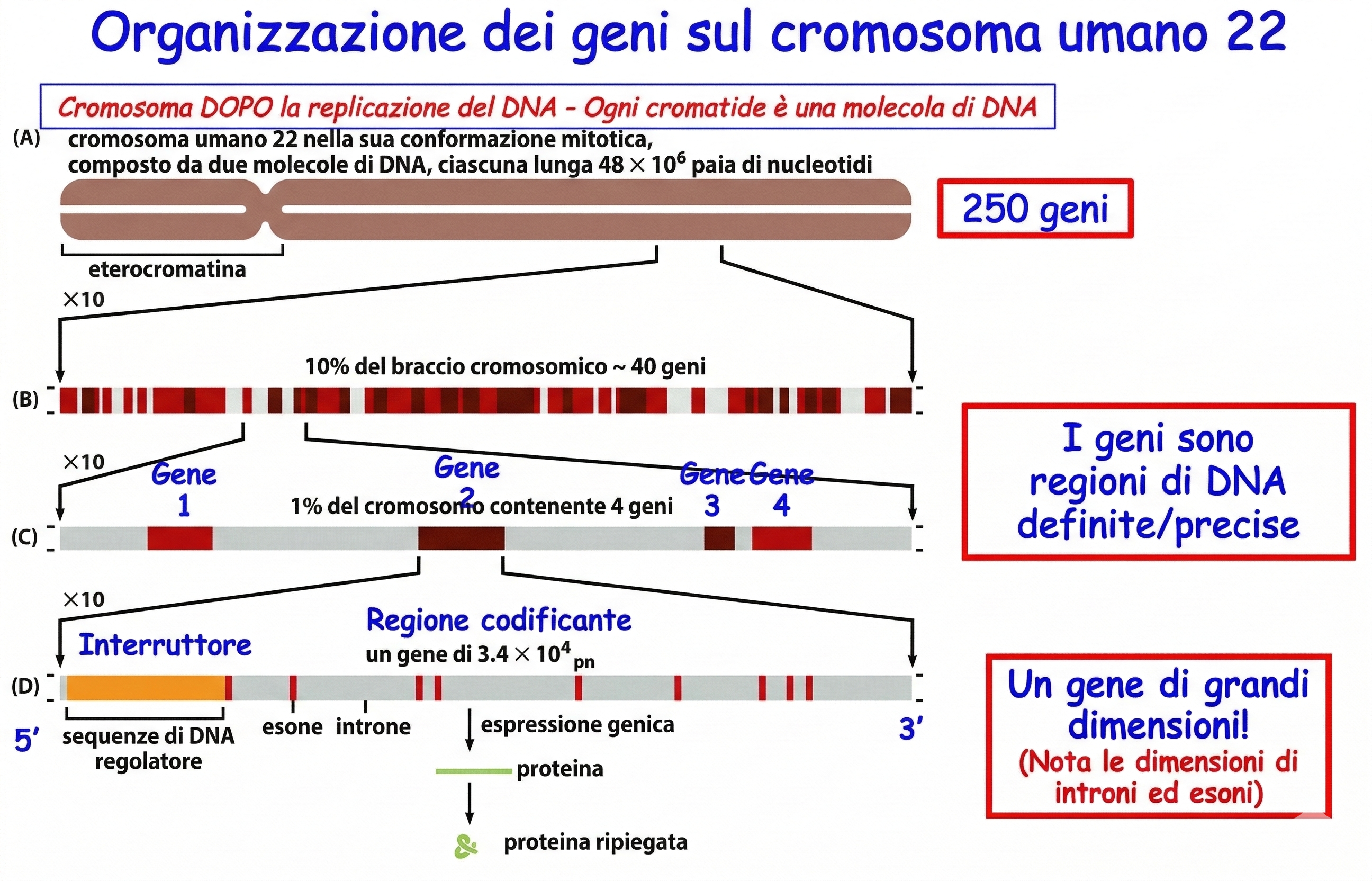
Il completamento della sequenza del cromosoma 22 nel 1999, uno dei più piccoli cromosomi umani, ha offerto per la prima volta una vista ad alta risoluzione dell’ordine e della densità dei geni lungo un intero cromosoma di vertebrato (Figura 04.05-02). Con la pubblicazione dell’intero genoma umano (bozza del 2001 e versione di riferimento del 2004) è stato possibile delineare un quadro globale dell’assetto genetico di tutti i cromosomi, includendo il numero di geni, le loro dimensioni e la loro distribuzione nei vari compartimenti genomici (Tabella 04.05-01). Un tratto che emerge con immediatezza è l’esiguità della frazione codificante per proteine, inferiore al 2% dell’intero genoma (Figura 04.05-03). Circa la metà del DNA è formata da elementi genetici mobili, testimoni delle ondate di colonizzazione che hanno interessato il nostro genoma durante l’evoluzione; la maggior parte di tali elementi è oggi inattiva a causa di mutazioni inattivanti accumulate nel tempo.
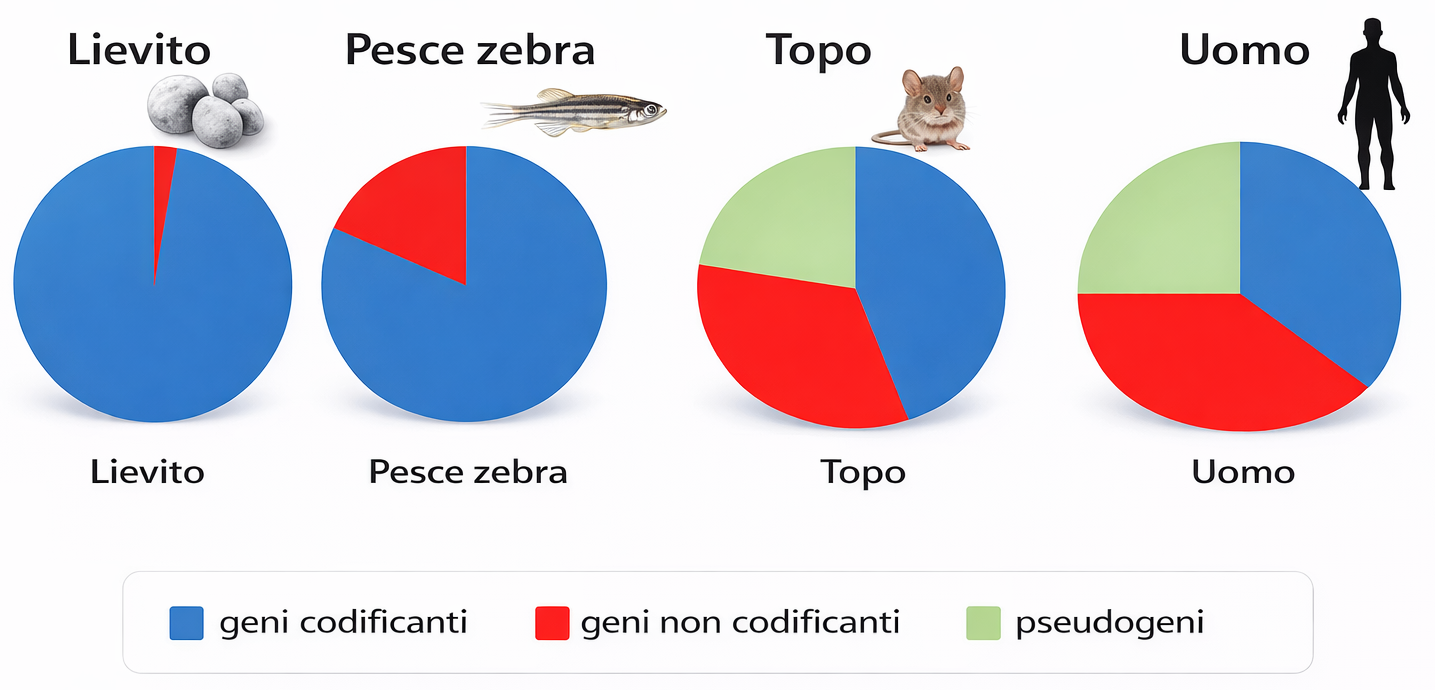
Anche il numero di geni che codificano proteine è più contenuto di quanto ipotizzato in origine. Stime iniziali di circa 100 000 geni si sono progressivamente ridimensionate: le valutazioni attuali indicano all’incirca 19 000 geni codificanti. A questi si aggiungono migliaia di loci che producono RNA funzionali non tradotti (ad esempio rRNA, tRNA e numerose classi di RNA non codificanti), per un ordine di grandezza di circa 5000 geni. Tali quantità non sono dissimili da quelle osservate in altri metazoi e piante modello, come Drosophila (circa 14 000), C. elegans (circa 22 000) e Arabidopsis (circa 28 000), suggerendo che la complessità di un organismo non dipende lineariamente dal mero conteggio dei geni, ma dall’elaborata regolazione e dalle combinazioni funzionali generate.
Pur essendo relativamente pochi, i geni umani sono mediamente voluminosi. Per codificare una proteina con circa 430 amminoacidi sono sufficienti circa \(430 \times 3 \approx 1{,}290\) nucleotidi nella regione codificante, ovvero approssimativamente 1300 coppie di basi. Tuttavia, la lunghezza media di un gene umano si aggira intorno a 27 000 coppie di basi, poiché gran parte della sequenza è occupata da introni e da regioni non tradotte. Oltre agli introni estesi (Figura 04.05-02), ciascun gene è corredato da un mosaico di sequenze regolatrici — promotori, enhancer, silencer, insulator — che orchestrano tempi, luoghi e livelli di espressione. Tali elementi sono spesso dispersi su decine di kilobasi e possono trovarsi sia a monte sia a valle del gene, talvolta anche a grande distanza lineare, riflettendo un’organizzazione tridimensionale del cromosoma in domini di interazione specifici. Nell’insieme, gli esoni e le sequenze regolatrici localmente associate rappresentano meno del 2% del genoma, rendendo quello umano nettamente meno compatto rispetto a molti altri genomi eucariotici (Figura 04.05-04).
Una sintesi della composizione genomica, utile a orientare il lettore, può essere espressa come segue:
- frazione codificante per proteine: < 2% del genoma totale;
- RNA non codificanti con funzione nota: migliaia di loci distribuiti in tutto il genoma;
- elementi mobili (LINE, SINE, LTR retrotrasposoni e DNA trasposoni): circa il 50% del genoma, per lo più non più attivi;
- introni e regioni intergeniche: la porzione predominante dello spazio genomico, con ampia variabilità di dimensioni e contenuti regolativi.
Il confronto tra specie e tra individui fornisce indizi sul “valore funzionale” delle sequenze. Gli studi di genomica comparata indicano che circa il 4,5% del genoma umano è altamente conservato tra i mammiferi, segno di vincoli selettivi nel tempo evolutivo. Analisi di variazione nella popolazione umana mostrano inoltre che un ulteriore 5% circa del genoma presenta variabilità ridotta, come rilevato dal confronto del DNA in campioni di migliaia di persone. Considerate insieme, tali osservazioni suggeriscono che approssimativamente il 10% del genoma sia sottoposto a vincoli funzionali rilevanti. Si tratta, tuttavia, di una stima di massima: molte regioni conservate non hanno ancora una funzione sperimentalmente definita, mentre porzioni meno conservate possono essere attive in specifici contesti cellulari o temporali. In sintesi, il genoma umano si distingue più per l’ampiezza del repertorio regolativo, la presenza di elementi ripetitivi e la modularità dell’espressione genica — con fenomeni come lo splicing alternativo e l’uso differenziale di promotori — che non per il semplice numero di geni. La “struttura del testo genetico” che ne deriva è ridondante e gerarchica, configurando un sistema capace di elevata plasticità funzionale e di adattamento, ma che al tempo stesso conserva tracce profonde della propria storia evolutiva.
Image Gallery
Statistiche fondamentali sul genoma umano
| Caratteristica del genoma umano | Valore indicativo |
|---|---|
| Lunghezza complessiva del DNA | ~3,2 × 10⁹ coppie di nucleotidi |
| Geni codificanti proteine | Circa 19.000 |
| Geni non codificanti (RNA funzionali) | ~5.000 |
| Numero stimato di pseudogeni | ~11.000 |
| Lunghezza massima di un gene | ~2,4 × 10⁶ coppie di nucleotidi |
| Lunghezza media di un gene | ~27.000 coppie di nucleotidi |
| Numero minimo di esoni per gene | 1 |
| Numero massimo di esoni per gene | 178 |
| Numero medio di esoni per gene | 10 |
| Dimensione massima di un esone | ~17.000 coppie di nucleotidi |
| Dimensione media di un esone | ~145 coppie di nucleotidi |
| Percentuale di DNA contenuto negli esoni | ~1,5% |
| Percentuale di DNA conservato rispetto ad altri mammiferi che non codifica proteine | ~3,5% |
| DNA presente in elementi ripetitivi ad alta copia | ~50% |
| Tabella che sintetizza le principali componenti del genoma umano, evidenziando la distribuzione tra regioni codificanti e non codificanti, la presenza di RNA funzionali, pseudogeni, sequenze ripetute e la ridotta quota rappresentata dagli esoni. | |
Image Gallery
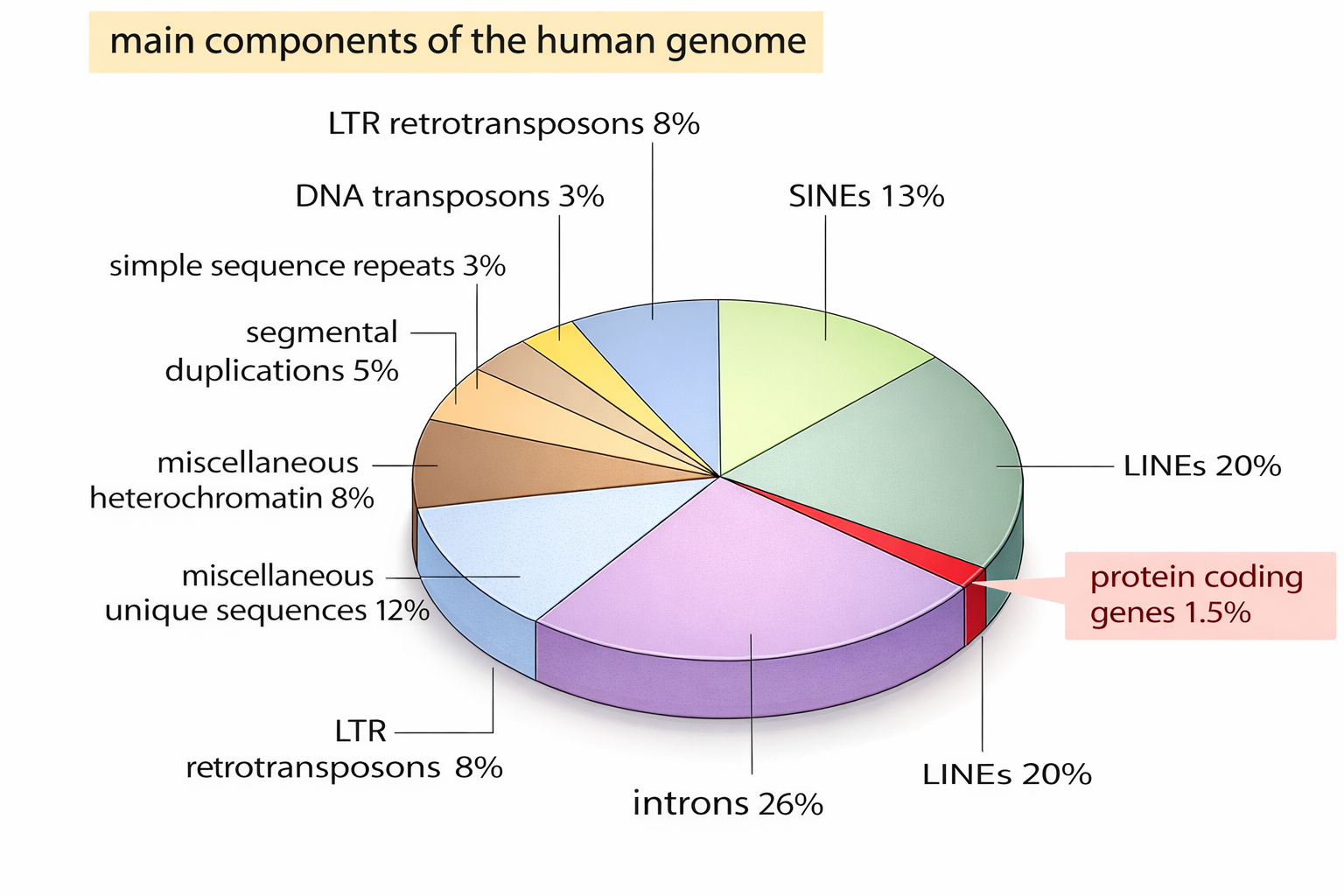
Image Gallery
Image Gallery
Le sequenze complete del genoma di numerosi mammiferi, tra cui esseri umani, scimpanzé, gorilla, oranghi, cani, gatti e topi, mostrano una sorprendente continuità: il repertorio di geni codificanti proteine è in gran parte condiviso. Da qui scaturiscono interrogativi centrali: se i geni sono pressoché gli stessi, da dove originano le marcate differenze morfologiche, fisiologiche e comportamentali tra specie? E quali basi molecolari distinguono gli esseri umani dagli altri animali? Una parte decisiva della risposta risiede nel DNA non codificante che governa l’espressione genica. Sequenze regolative cis, spesso distribuite su ampie regioni genomiche, contengono insiemi di brevi siti di legame per fattori di trascrizione che, combinandosi, coordinano il quando, il dove e il quanto un gene viene trascritto.
Promotori, enhancers, silencers e insulator, integrati in moduli regolativi, orchestrano programmi spaziotemporali di espressione che guidano lo sviluppo embrionale e la specializzazione cellulare. La “logica” di questi moduli è resa possibile dall’interazione con la cromatina e dall’architettura tridimensionale del genoma, in cui domini topologicamente associati e anse di cromatina mettono in contatto fisico enhancers e promotori distanti. Per tali ragioni, l’evoluzione dei caratteri morfologici e fisiologici tende a riflettere più spesso modifiche nelle sequenze regolative e nella loro organizzazione che variazioni nelle sequenze proteiche o negli RNA funzionali.
Un approccio efficace per individuare tratti distintivi della nostra specie consiste nel cercare sequenze regolative altamente conservate in molti mammiferi ma alterate o assenti nell’uomo. Analisi comparative hanno identificato centinaia di tali regioni, rivelando modalità con cui piccole differenze regolative possano avere effetti fenotipici sostanziali. Un esempio proviene da una regione regolativa assente nel genoma umano che, in altre specie, limita la proliferazione neuronale in aree cerebrali in sviluppo; la sua perdita, o modifiche in elementi affini attivi nei progenitori neurali, potrebbe aver contribuito all’espansione e alla riorganizzazione del cervello umano. Un’altra regione, ugualmente assente nell’uomo, controlla la formazione delle spine peniene, strutture diffuse in molti primati, inclusi scimpanzé, bonobo, gorilla, oranghi e gibboni. Non è chiaro se la perdita di tali strutture abbia comportato un vantaggio selettivo o sia stata neutra; in ogni caso, rappresenta una peculiarità della nostra specie.
Questi confronti indicano che variazioni nel repertorio e nell’attività degli enhancers, insieme a cambiamenti strutturali che possono ricollocare elementi regolativi rispetto ai loro geni bersaglio, hanno avuto un ruolo chiave nella divergenza tra specie. Rimane tuttavia una distinzione temporale: tali analisi illuminano soprattutto le tappe antiche della nostra storia evolutiva. Per eventi più recenti, risultano decisive le informazioni derivate dallo studio dei genomi delle specie estinte a noi più prossime.
Nel 2010 è stata pubblicata la prima analisi di ampio respiro del genoma dei Neanderthal, i parenti più prossimi degli umani moderni. Il DNA, estratto da resti fossili rinvenuti in una grotta in Croazia, ha consentito un confronto sistematico con i genomi di individui contemporanei provenienti da diverse aree geografiche. Questo confronto ha messo in evidenza un insieme di regioni umane che hanno accumulato cambiamenti con una rapidità insolitamente elevata, incluse aree che coinvolgono geni legati al metabolismo, allo sviluppo e al rimodellamento del cervello, a strutture della laringe e alla conformazione dello scheletro, con particolare riferimento a gabbia toracica e regione frontale, tratti nei quali si ritengono esistere differenze tra umani moderni e Neanderthal. Un risultato inatteso e di grande portata è la presenza, in una quota significativa di individui odierni di origine europea e asiatica, di segmenti di ascendenza neandertaliana che rappresentano circa il 2% del genoma. Questa eredità indica che, dopo la dispersione degli umani moderni fuori dall’Africa, si verificarono episodi di incrocio con i Neanderthal, prima della scomparsa di questi ultimi (Figura 04.05-05). I segmenti di origine arcaica sono distribuiti a mosaico lungo il genoma e, in alcuni casi, hanno subito selezione positiva o negativa in funzione del loro effetto biologico: si sono osservati contributi a vie immunitarie e a caratteri cutanei e piliferi, nonché deplezione in regioni associate alla spermatogenesi, verosimilmente per incompatibilità funzionali. Nel loro insieme, queste evidenze mostrano che una frazione della variabilità genetica contemporanea deriva dall’intreccio tra linee evolutive umane distinte, e che tale intreccio ha lasciato un’impronta permanente, funzionalmente rilevante in alcune circostanze, sul genoma della nostra specie.
Image Gallery

Tranne nel caso dei gemelli monozigoti, ogni persona possiede una combinazione irripetibile di varianti genetiche. Confrontando la stessa regione genomica in due individui qualsiasi si osserva, in media, una differenza pari allo 0,1% della sequenza nucleotidica, cioè circa 1 variazione ogni 1000 coppie di basi. Considerando la dimensione del genoma umano, ciò si traduce in all’incirca 3 milioni di differenze tra due genomi. Una parte sostanziale di tale diversità è antica e risale a centinaia di migliaia di anni fa, quando l’antenata popolazione umana aveva dimensioni ridotte; ciascun individuo moderno eredita combinazioni uniche di queste varianti storiche. A queste si sommano mutazioni de novo, introdotte ad ogni generazione: alla nascita, ciascun genoma contiene tipicamente 60–80 mutazioni nuove, non presenti nei genitori, in accordo con un tasso di mutazione di circa \(1,0 \times 10^{-8}\) per base per generazione.
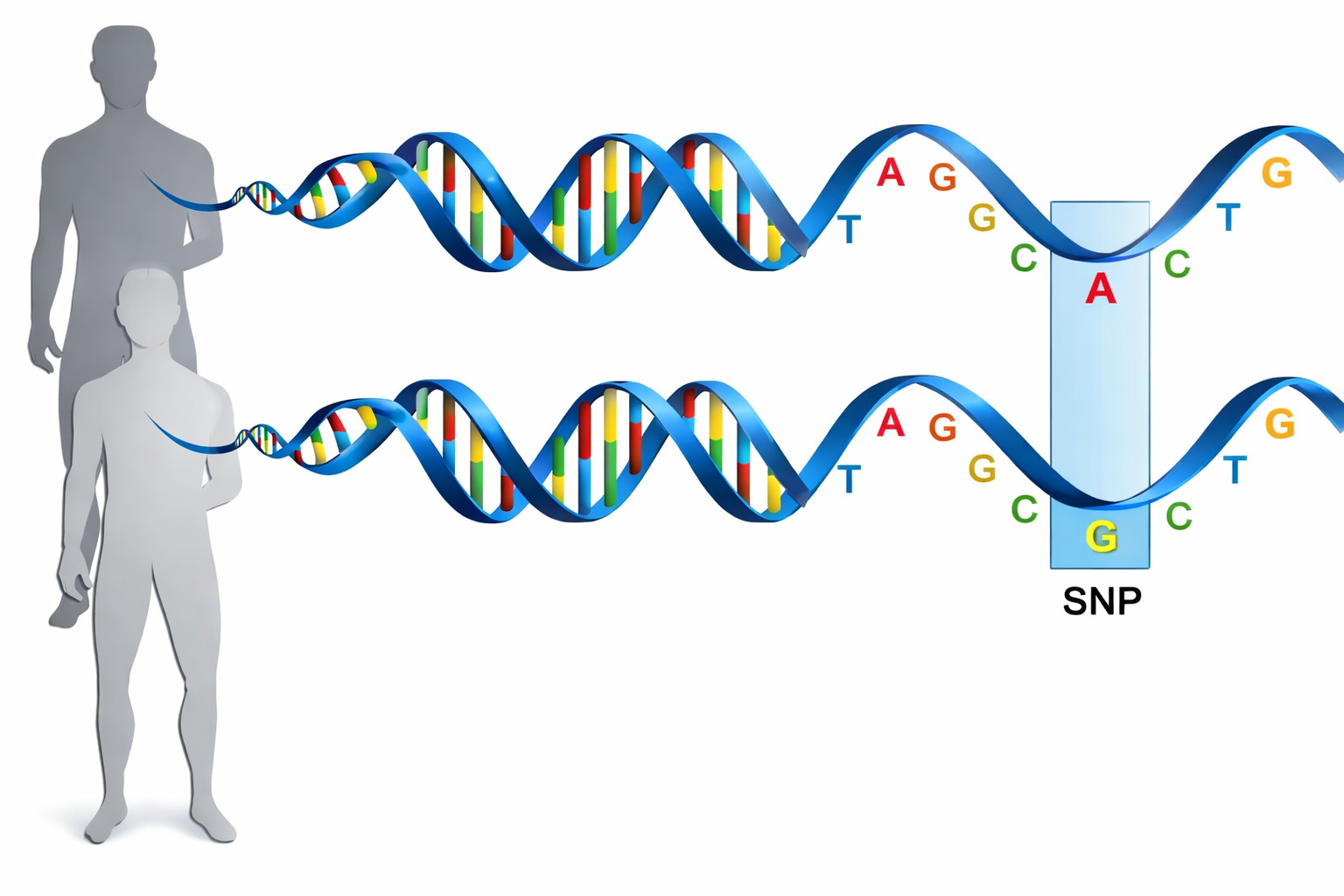
La frazione più grande della variabilità è costituita da sostituzioni puntiformi. Quando una variante a singola base è presente con frequenza almeno dell’1% nella popolazione, viene definita polimorfismo a singolo nucleotide (SNP). Due genomi umani scelti casualmente differiscono in media per circa \(2,5 \times 10^6\) SNP distribuiti sull’intero genoma (Figura 04.05-06). Oltre agli SNP, sono comuni altre classi di varianti, tra cui piccole inserzioni/delezioni (indel) e variazioni del numero di copie (CNV) che coinvolgono segmenti anche di grandi dimensioni, capaci di modificare l’assetto regolativo e l’architettura 3D del genoma:
- molti SNP sono neutri dal punto di vista funzionale, soprattutto se collocati in regioni prive di ruoli regolativi o codificanti;
- un sottoinsieme cade in esoni, splice sites o elementi regolativi, con effetti potenzialmente misurabili su proteine, trascrizione ed epigenetica;
- varianti non codificanti possono alterare l’affinità di legame dei fattori di trascrizione, modulando l’espressione in modo cellula-specifico attraverso eQTL;
- la correlazione non casuale tra varianti contigue (disequilibrio di collegamento) genera aplotipi che facilitano l’identificazione di associazioni tra regioni genomiche e tratti complessi tramite studi su larga scala.
Benché la gran parte delle differenze non influenzi il fenotipo, una quota minoritaria contribuisce a tratti ereditabili. Un esempio classico è la persistenza della lattasi in età adulta, dovuta a varianti regolative in un enhancer remoto del gene LCT. Molti tratti complessi, inclusi caratteri fisiologici e risposta ai farmaci, sono modulati da insiemi di varianti ciascuna con effetto modesto, spesso situate in enhancers attivi nei tessuti pertinenti. Le interazioni tra varianti genetiche, ambiente e microbiota aggiungono ulteriori livelli di complessità, spiegando parte delle differenze interindividuali in suscettibilità alle malattie e negli esiti terapeutici. In definitiva, la nostra individualità biologica scaturisce dalla combinazione di antiche varianti condivise, mutazioni recenti e riassetti regolativi che modulano reti di espressione genica. Comprendere come queste componenti si integrino per produrre differenze tra individui rimane una delle sfide più ambiziose per la biologia molecolare e cellulare.
Image Gallery