Gli atomi di carbonio possono formare molecole diverse legandosi ad altri quattro atomi
TOPICS
Definizione

Le proteine sono macromolecole biologiche straordinariamente articolate sia nella struttura sia nella funzione. Dal punto di vista chimico, esse sono polimeri di amminoacidi che, una volta assemblati in catene polipeptidiche, si ripiegano in architetture tridimensionali specifiche stabilizzate prevalentemente da interazioni non covalenti tra porzioni distinte della stessa catena. L’evoluzione ha affinato nel tempo sia l’assetto strutturale sia le proprietà funzionali di ciascuna proteina, dando luogo a un’ampia varietà di forme e attività. La conoscenza della struttura a risoluzione atomica permette di comprendere come la conformazione determini la funzione nel contesto cellulare, in accordo con il principio secondo cui la forma è intimamente connessa all’attività biologica. Nel descrivere le proteine, è utile distinguere tra: organizzazione primaria (sequenza amminoacidica), secondaria (motivi locali, come α-eliche e foglietti β), terziaria (ripiegamento complessivo della singola catena) e quaternaria (assemblaggio di più subunità), livelli che emergono dalla stessa chimica del polipeptide e dal suo ambiente.
Le proteine sono costruite impiegando principalmente 20 amminoacidi standard, ciascuno dotato di proprietà fisico-chimiche distintive. Ciascuna catena è formata da amminoacidi uniti da legami peptidici covalenti che collegano il gruppo carbossilico di un residuo al gruppo amminico del successivo (Figura 01.16-01). Per questo motivo le proteine sono dette anche polipeptidi, e le loro catene, catene polipeptidiche. La sequenza amminoacidica, unica per ogni proteina, è rigorosamente conservata per quella specifica molecola; ad esempio, tutte le molecole di emoglobina umana condividono la medesima sequenza per ciascuna delle loro subunità.
L’ossatura polipeptidica è una ripetizione regolare di atomi disposti secondo lo schema –N–C–C–, comune a tutti gli amminoacidi (Figura 01.16-02). Le due estremità della catena sono chimicamente distinte: l’estremo con il gruppo amminico libero è detto N-terminale, quello con il gruppo carbossilico libero è il C-terminale. Dall’ossatura sporgono le catene laterali (gruppi R), non coinvolte direttamente nel legame peptidico, responsabili delle proprietà peculiari di ciascun amminoacido (Figura 01.16-02). Alcune catene laterali sono apolari e idrofobe, altre polari o cariche, altre ancora particolarmente reattive. La (Figura 01.16-03) riassume struttura e denominazioni dei 20 amminoacidi proteici.
Le catene polipeptidiche sono intrinsecamente flessibili perché diversi legami a singolo legame dell’ossatura consentono rotazioni dei diedri attorno all’atomo di Cα; tali rotazioni, note come angoli φ (phi) e ψ (psi), definiscono lo spazio conformazionale accessibile. Non tutte le combinazioni sono però permesse: vincoli sterici e preferenze energetiche, sintetizzate dal diagramma di Ramachandran, limitano le conformazioni adottabili. In questo contesto, la forma finale è selezionata e mantenuta da una rete di interazioni deboli che coinvolgono sia l’ossatura sia le catene laterali:
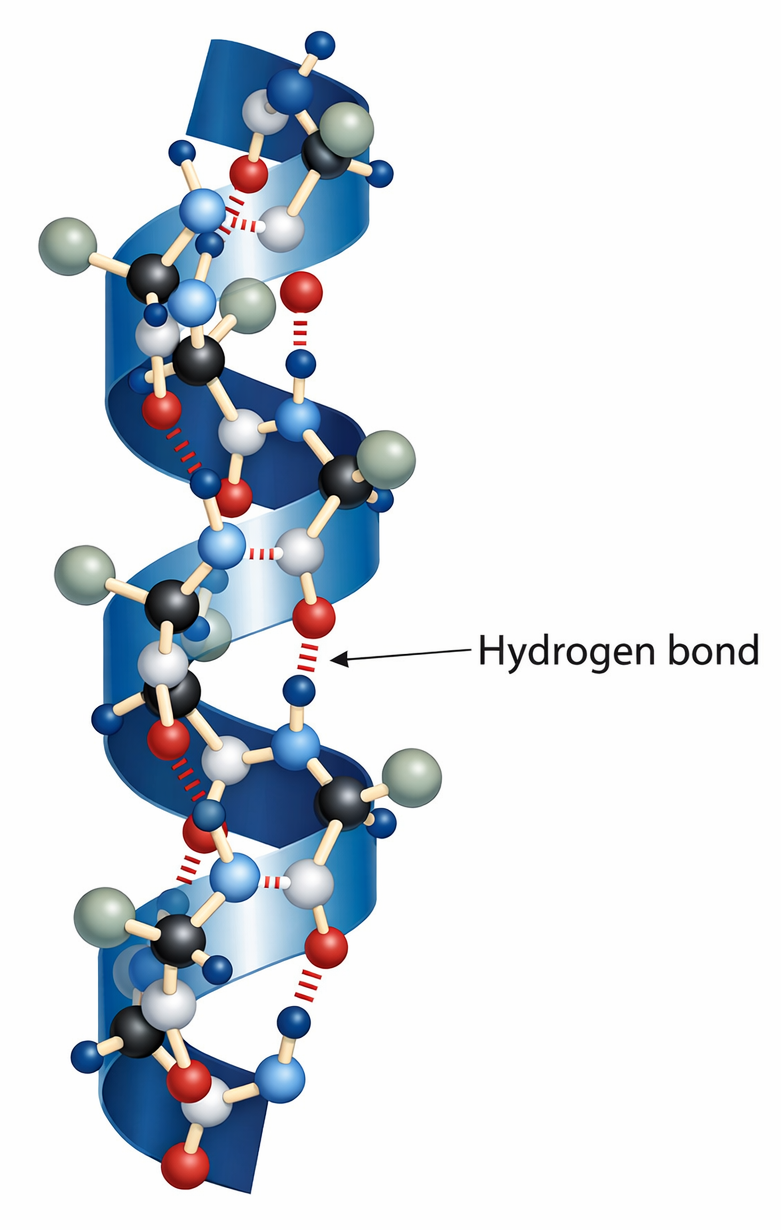
- legami a idrogeno, che stabilizzano motivi locali (come α-eliche e foglietti β) e contatti a lungo raggio;
- interazioni elettrostatiche (incluse coppie ioniche o “salt bridge”) tra residui carichi di segno opposto;
- forze di van der Waals, dovute a interazioni dipolari transitorie a corto raggio;
- effetto idrofobico, che promuove il raggruppamento di catene laterali apolari in regioni schermate dall’acqua.
Poiché ciascuna di queste interazioni ha forza individuale modesta rispetto a un legame covalente, la stabilità complessiva della conformazione dipende dall’azione combinata di molti contatti cooperativi (Figura 01.16-04). Un contributo cruciale deriva dall’effetto idrofobico: in ambiente acquoso, i residui apolari tendono a segregare dall’acqua e a concentrarsi nel cuore della proteina ripiegata, minimizzando la perturbazione della rete di legami a idrogeno del solvente. Residui come leucina, valina, fenilalanina e triptofano (Figura 01.16-03) si localizzano perciò tipicamente all’interno, mentre residui polari o carichi, come arginina, glutammina e istidina, si dispongono verso la superficie dove possono formare legami a idrogeno e interazioni elettrostatiche con l’acqua e con altre molecole polari (Figura 01.16-05). Quando residui polari si trovano all’interno, di frequente stabiliscono legami a idrogeno con altri gruppi polari o con l’ossatura, contribuendo ulteriormente alla stabilità (Figura 01.16-06). In aggiunta alle forze non covalenti, in talune proteine possono formarsi ponti disolfuro tra residui di cisteina, che fungono da “cravatte” covalenti stabilizzanti in ambienti ossidanti.
Image Gallery

Image Gallery
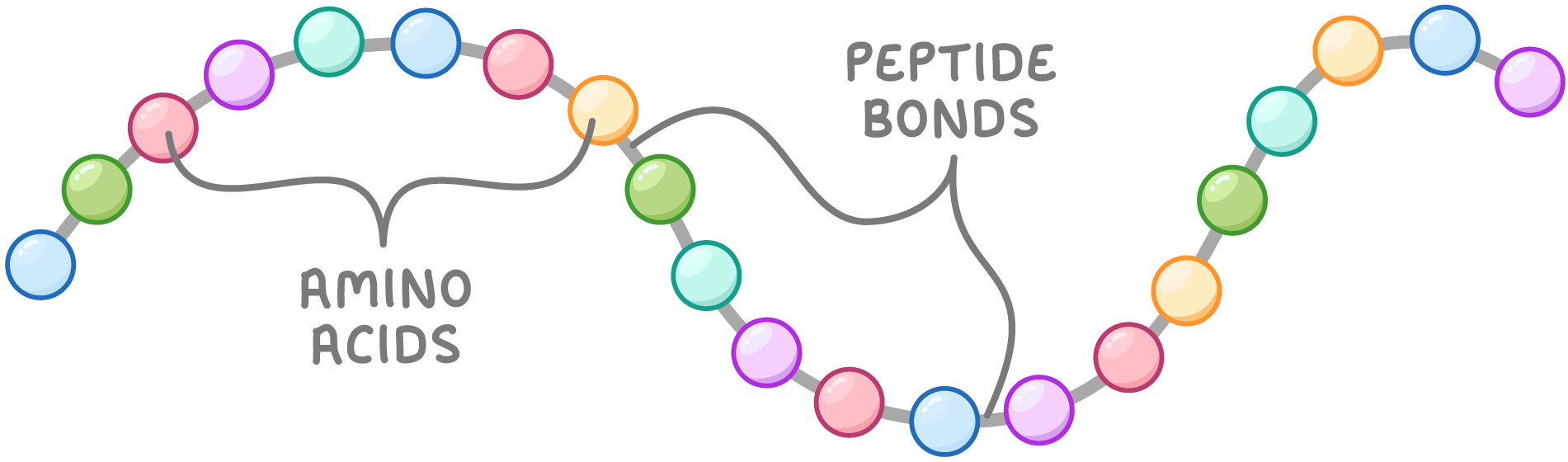
Image Gallery
Image Gallery

Image Gallery

Image Gallery
Image Gallery
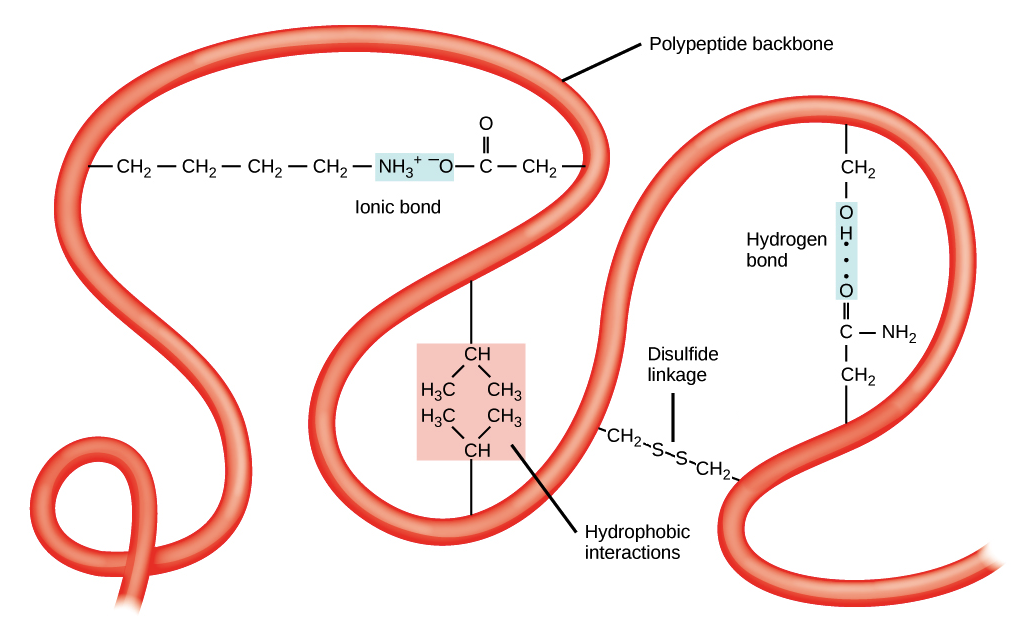
Image Gallery
Ogni proteina tende a ripiegarsi nella conformazione tridimensionale favorita termodinamicamente dalla propria sequenza amminoacidica. La stabilità relativa delle conformazioni è governata dall’energia libera di Gibbs, riassunta dall’espressione \( \Delta G = \Delta H - T \Delta S \). In acqua, il ripiegamento bilancia contributi entalpici (numerose interazioni specifiche all’interno della proteina) e contributi entropici: se da un lato la catena perde libertà conformazionale, dall’altro il solvente guadagna entropia quando i residui idrofobi vengono sequestrati dal nucleo proteico. Nel complesso, la conformazione nativa corrisponde a un minimo locale (spesso globale) di \( G \) e il processo risulta favorito, con rilascio di calore e incremento del disordine del sistema complessivo proteina–solvente.
Queste considerazioni sono state corroborate da studi su proteine purificate. L’applicazione di agenti denaturanti in grado di rompere le interazioni non covalenti, come urea o cloridrato di guanidinio, o di agenti riducenti per scindere ponti disolfuro, induce la perdita della struttura nativa e produce catene flessibili prive di attività specifica. Rimuovendo i denaturanti e ripristinando condizioni fisiologiche, molte proteine recuperano spontaneamente la loro conformazione originaria, fenomeno noto come rinaturazione (Figura 01.16-07). Ciò dimostra che l’informazione necessaria a specificare la struttura tridimensionale è codificata nella sequenza primaria.
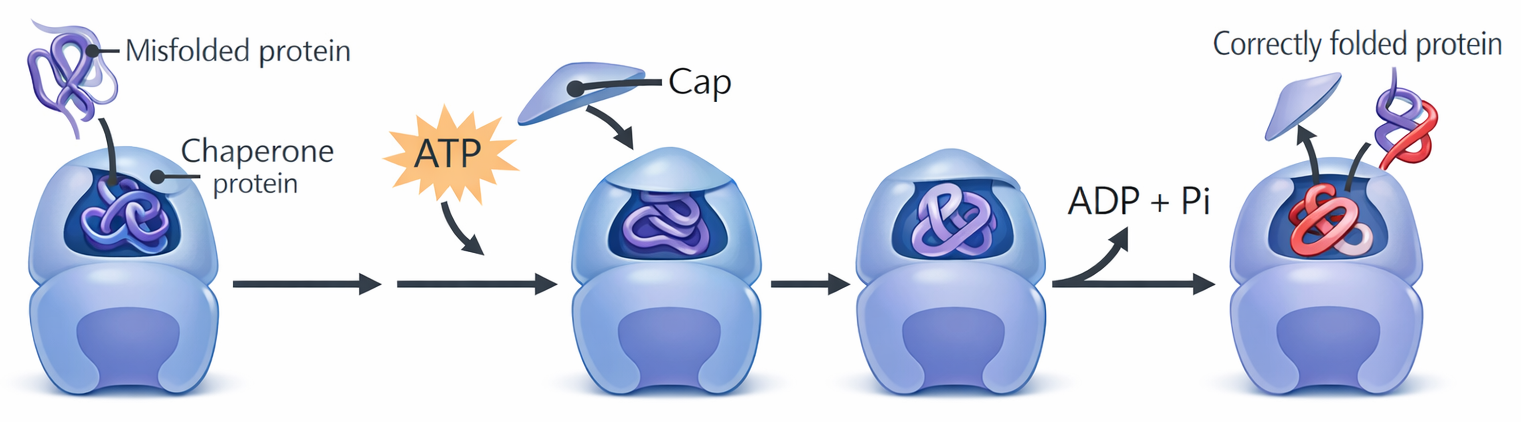
Nell’ambiente cellulare, ricco di macromolecole e potenziali superfici d’aggregazione, il ripiegamento è assistito da proteine chaperon. Alcune si legano transitoriamente a catene nascenti o parzialmente ripiegate, impedendo interazioni indesiderate e facilitando l’accesso al percorso energeticamente più favorevole (Figura 01.16-08); altre, dette chaperonine, forniscono vere e proprie “camere di isolamento” in cui la catena può ripiegarsi lontano dal rischio di aggregazione (Figura 01.16-09). In molte di queste reazioni l’idrolisi di ATP dirige cicli di legame e rilascio che aumentano l’efficienza del processo. È importante sottolineare che le chaperon non determinano la struttura finale: la conformazione nativa resta specificata dalla sequenza; esse accelerano e rendono più affidabile il raggiungimento di tale stato.
Benché per la maggior parte delle proteine la conformazione nativa sia ben definita e stabile, piccole variazioni indotte dall’interazione con ligandi, altre proteine o modificazioni post-traduzionali possono modulare in modo regolato la struttura e, di conseguenza, la funzione. Queste dinamiche conformazionali, alla base di fenomeni come l’allosteria e l’induced fit, sono essenziali per l’attività catalitica degli enzimi, la trasduzione del segnale e il riconoscimento molecolare. D’altra parte, la presenza di stati meta-stabili e di “trappole” nell’energia paesaggistica del ripiegamento può, in condizioni sfavorevoli, sfociare in misfolding e aggregazione; la cellula limita tali eventi grazie alla sorveglianza delle chaperon e a sistemi di qualità proteica.
Image Gallery
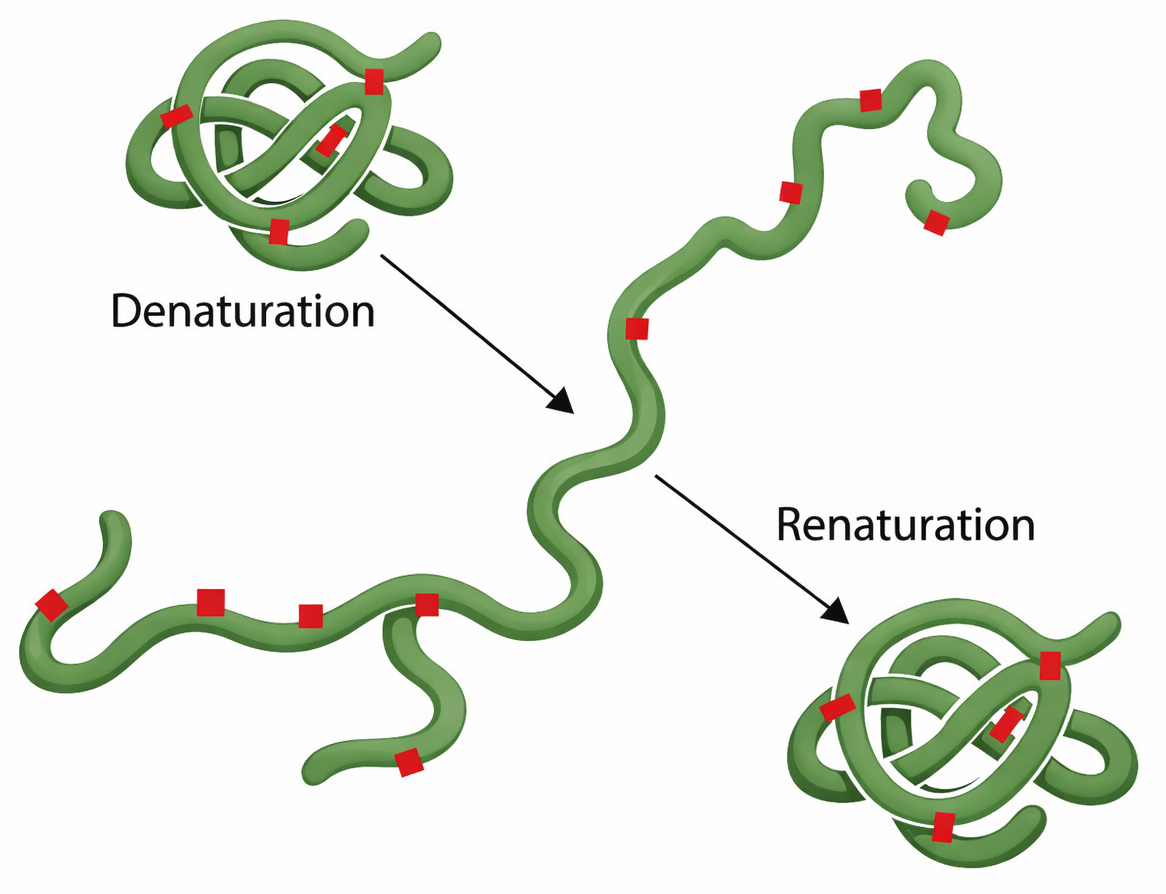
Image Gallery

Image Gallery
Tra le macromolecole cellulari, le proteine sono quelle con la più ampia diversità strutturale. Le catene polipeptidiche possono essere molto corte, circa 30 amminoacidi, oppure estremamente lunghe, superando le 10 000 unità; la maggior parte rientra comunque nell’intervallo 50–2000 residui. Le architetture spaziali comprendono conformazioni globulari e fibrose; si organizzano in filamenti, fogli, anelli e strutture quasi sferiche (Figura 01.16-10). Strutture di questo tipo ricorreranno più volte nelle pagine che seguono. Sfruttando tecniche strutturali complementari, sono state caratterizzate ormai oltre 100 000 proteine, e il numero continua a crescere con il contributo di diffrazione a raggi X, criomicroscopia elettronica e risonanza magnetica nucleare, insieme ai principali repertori internazionali (per esempio il Protein Data Bank).
La maggior parte delle proteine adotta una conformazione tridimensionale irregolare e intricata, spesso difficilmente descrivibile in modo esaustivo. Per intuire la complessità di tali conformazioni può essere utile osservare una proteina di dimensioni ridotte, come la proteina batterica di trasporto HPr, formata da soli 88 amminoacidi e implicata nel trasferimento di zuccheri nelle cellule batteriche. La sua struttura tridimensionale è rappresentata in quattro modalità nella (Figura 01.16-11), ciascuna utile a mettere in evidenza aspetti differenti:
- tracciato dell’ossatura polipeptidica (Figura 01.16-11), che fornisce la mappa generale del percorso della catena e consente un confronto diretto tra proteine imparentate;
- modello a nastro (Figura 01.16-11), che enfatizza gli elementi di struttura secondaria e i loro ripiegamenti;
- rappresentazione a bastoncini o “a filo ramificato” (Figura 01.16-11), utile a localizzare tutte le catene laterali e a inferire quali residui possano partecipare alla funzione;
- modello a spazio pieno (Figura 01.16-11), che delinea la superficie accessibile ai solventi, evidenziando i residui esposti e il modo in cui la proteina interagisce con piccole molecole o con altre macromolecole.
All’aumentare delle dimensioni proteiche, o quando più subunità si associano in complessi, la visualizzazione richiede strumenti grafici e algoritmi che consentano rotazioni, zoom e sezionamenti, come illustrato in (Figura 01.16-11). Il confronto sistematico di strutture tridimensionali evidenzia che, pur essendo ogni architettura globale unica, ricorrono motivi conformazionali preferenziali, come discussi nei paragrafi successivi.
Image Gallery
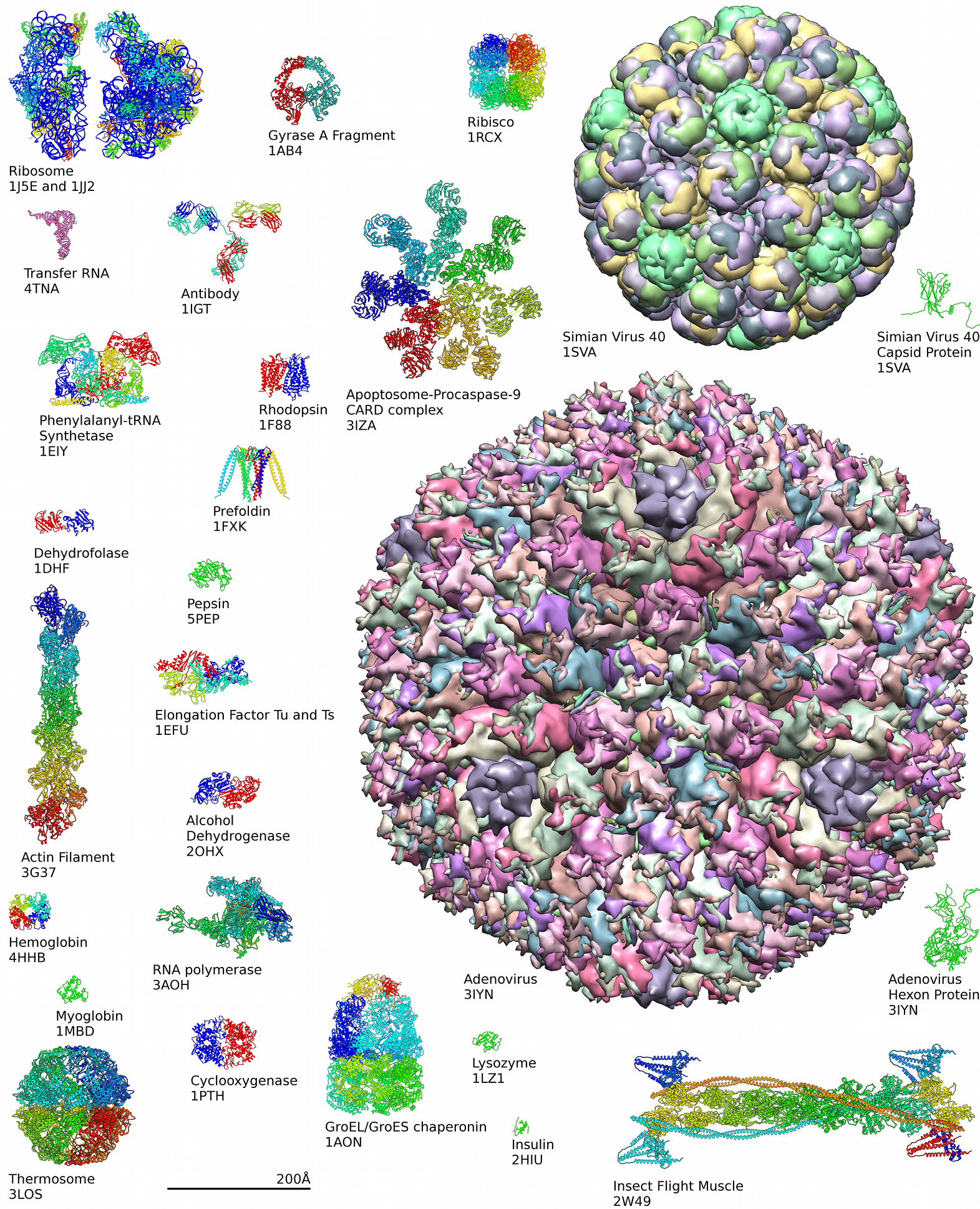
Image Gallery
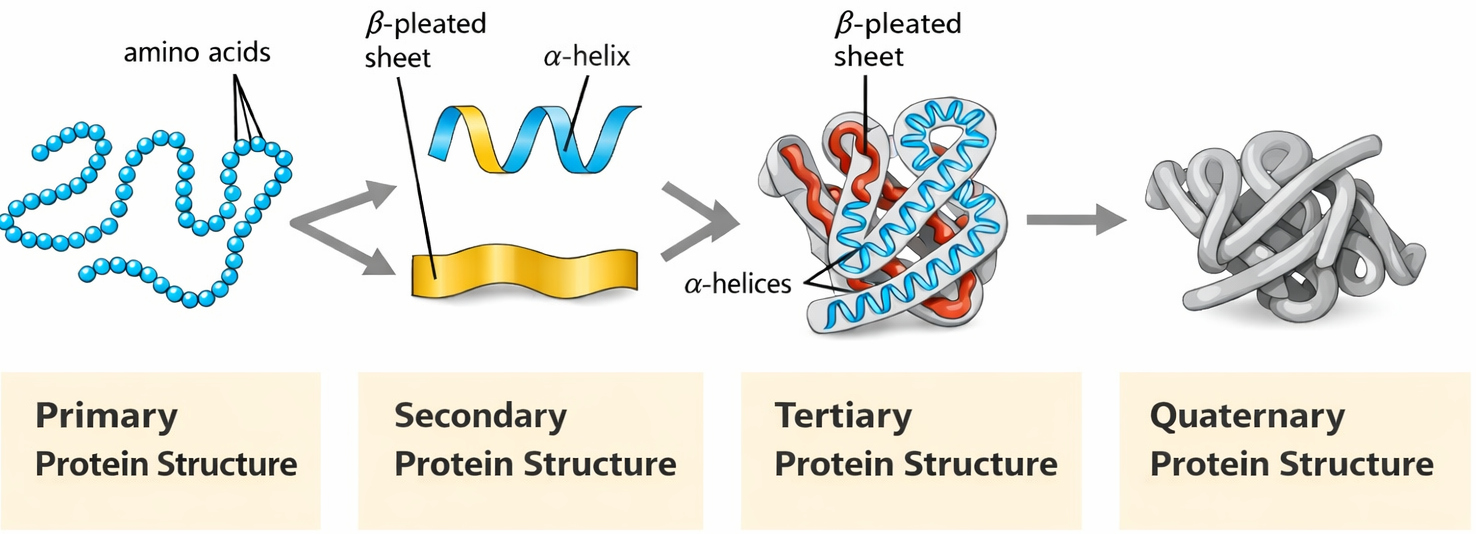
Image Gallery
Image Gallery
Image Gallery
Image Gallery
Image Gallery
Analisi classiche di materiali biologici come capelli e seta, condotte oltre mezzo secolo fa, portarono all’individuazione di due modalità di ripiegamento condivise da numerose proteine. La prima, l’α‑elica, venne identificata nell’α‑cheratina, proteina abbondante nell’epidermide e nei suoi derivati (peli, unghie, corna). Poco dopo fu riconosciuto un secondo motivo, il foglietto β, nella fibroina, componente fondamentale della seta. L’adozione di lettere greche riflette l’ordine storico delle scoperte.
Entrambe le conformazioni sono favorite dalla formazione di legami a idrogeno tra i gruppi N–H e C=O dell’ossatura peptidica (Figura 01.16-06). Poiché questi legami coinvolgono la dorsale polipeptidica e non le catene laterali, sequenze amminoacidiche diverse possono generare α‑eliche o foglietti β, producendo pattern regolari e ripetitivi. Le caratteristiche geometriche di tali motivi e le convenzioni grafiche con cui vengono rappresentati nei modelli sono sintetizzate nelle (Figura 01.16-12) e (Figura 01.16-13). Considerazioni steriche, spesso visualizzate nel diagramma di Ramachandran, indicano le regioni angolari consentite per i legami peptidici coerenti con α‑eliche e foglietti β.
Image Gallery
Image Gallery
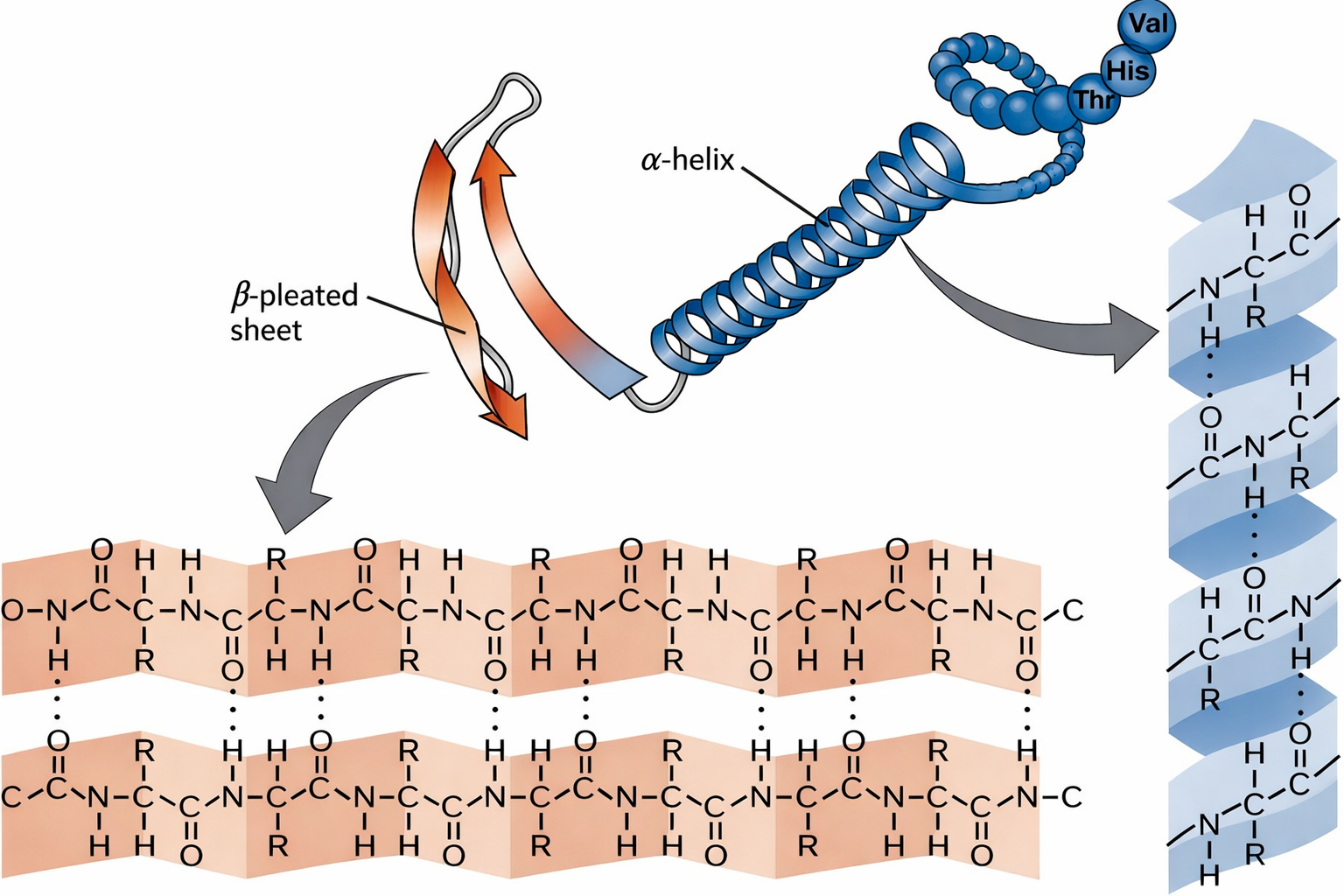
Image Gallery

La forma elicoidale è comune in natura perché corrisponde a un’organizzazione regolare ottenuta allineando unità simili in modo sfalsato, analogamente a una scala a chiocciola (Figura 01.16-14). La chiralità distingue eliche destre e sinistre (Figura 01.16-14): il verso non muta capovolgendo l’elica, mentre si inverte con una riflessione allo specchio.
Nell’α‑elica, la catena polipeptidica si avvolge su sé stessa formando un cilindro compatto stabilizzato da legami a idrogeno intrafilamento tra il gruppo C=O del residuo i e il gruppo N–H del residuo i+4 (Figura 01.16-12). La geometria risultante presenta \(3{,}6\) residui per giro e un passo di circa \(5{,}4\) Å per avvolgimento, con un’andatura normalmente destrosa nelle proteine naturali. La stabilità dell’elica è modulata dalla natura delle catene laterali, che possono favorire o ostacolare l’impaccamento locale.
Tratti elicoidali compaiono con alta frequenza nelle proteine di membrana. Nel passaggio attraverso il doppio strato fosfolipidico, segmenti transmembrana adottano tipicamente la conformazione di α‑elica e mostrano un arricchimento in residui idrofobi; in tal modo, l’ossatura polipeptidica, polare, soddisfa internamente i propri legami a idrogeno, mentre le catene laterali apolari schermano l’elica dall’ambiente lipidico (Figura 01.16-15). Questo principio è cruciale per recettori, canali e trasportatori integrali di membrana.
Quando due o più α‑eliche mostrano facce idrofobe complementari, possono avvolgersi l’una sull’altra dando origine a strutture superavvolte (coiled‑coil), particolarmente stabili (Figura 01.16-16). Una caratteristica frequente in tali segmenti è il motivo a eptade, con posizioni a e d preferenzialmente idrofobiche, che promuove l’associazione elica‑elica e riduce l’esposizione al solvente acquoso. Le eliche superavvolte costituiscono il nucleo meccanico di proteine allungate come l’α‑cheratina, impalcatura delle fibre intracitoplasmatiche dell’epidermide, e la miosina, motore della contrazione muscolare.
Image Gallery
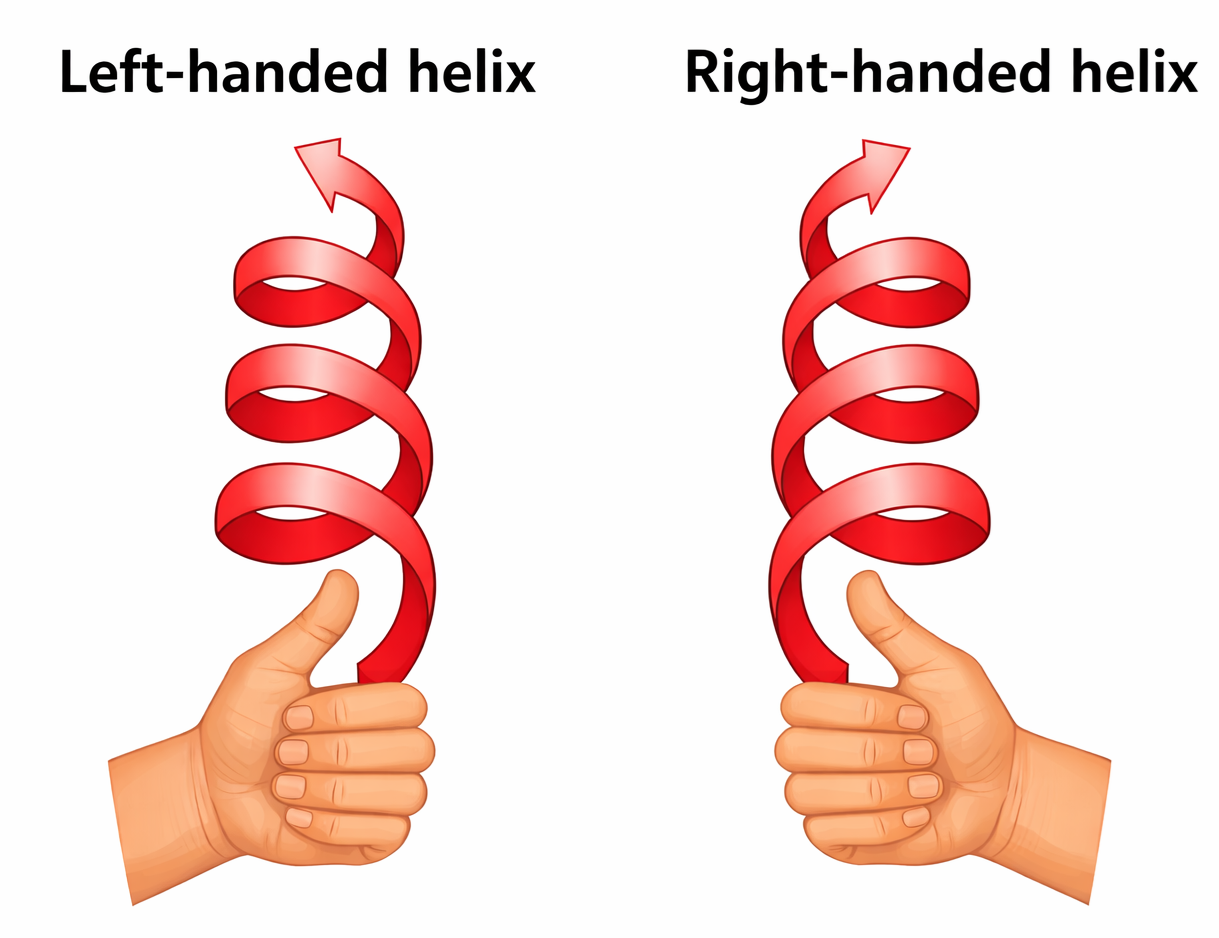
Image Gallery
Image Gallery
Image Gallery
Image Gallery

I foglietti β emergono quando segmenti di catena polipeptidica affiancati si connettono tramite legami a idrogeno tra ossature adiacenti (Figura 01.16-13). Se i filamenti scorrono nella stessa direzione sequenziale (N→C), il foglietto è detto parallelo; se invece la direzione è opposta tra filamenti vicini, si parla di antiparallelo (Figura 01.16-17). Entrambi danno origine a superfici pieghettate, meccanicamente rigide, che spesso contribuiscono al “core” idrofobo delle proteine. Anche la piccola HPr (Figura 01.16-11) contiene più filamenti β organizzati in un foglietto compatto.
I foglietti β impartiscono proprietà fisiche peculiari. La fibroina della seta deve la sua notevole resistenza alla trazione all’impaccamento regolare di foglietti β estesi. Inoltre, questi motivi consentono la formazione di fibre amiloidi: aggregati poco solubili stabilizzati da impilamenti di foglietti β con le catene laterali che si interdigitano come i denti di una cerniera (Figura 01.16-18). Tali architetture “cross‑β” possono svolgere funzioni cellulari specifiche, ma in alcuni contesti risultano coinvolte nel peggioramento di patologie proteotossiche.
Image Gallery
Image Gallery
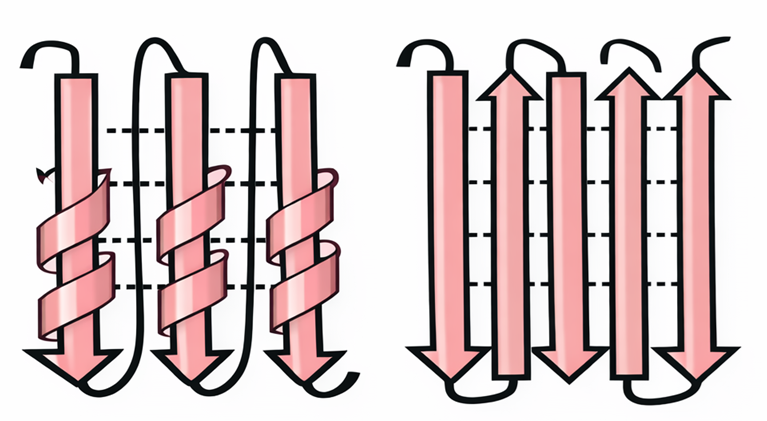
Image Gallery
Image Gallery
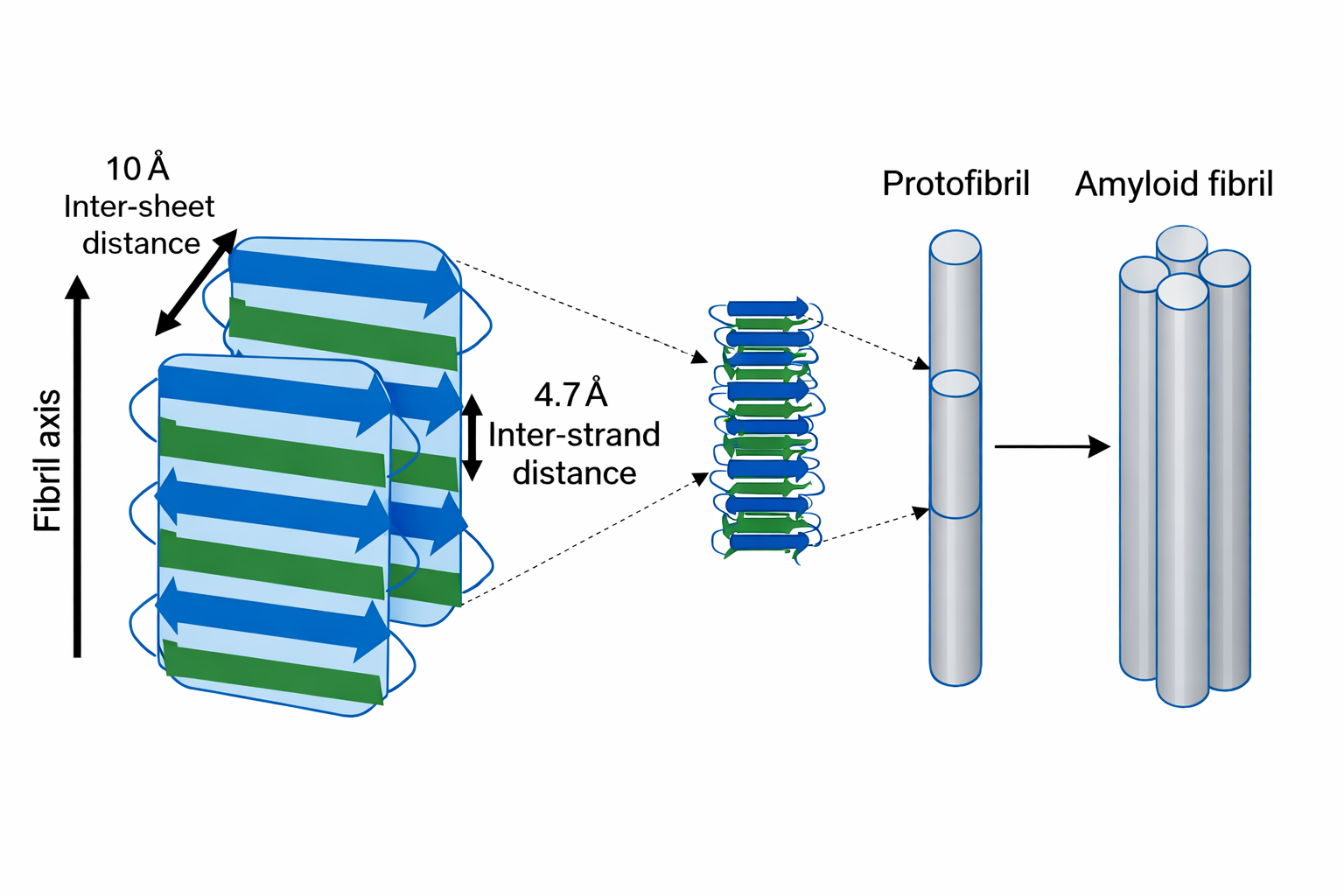
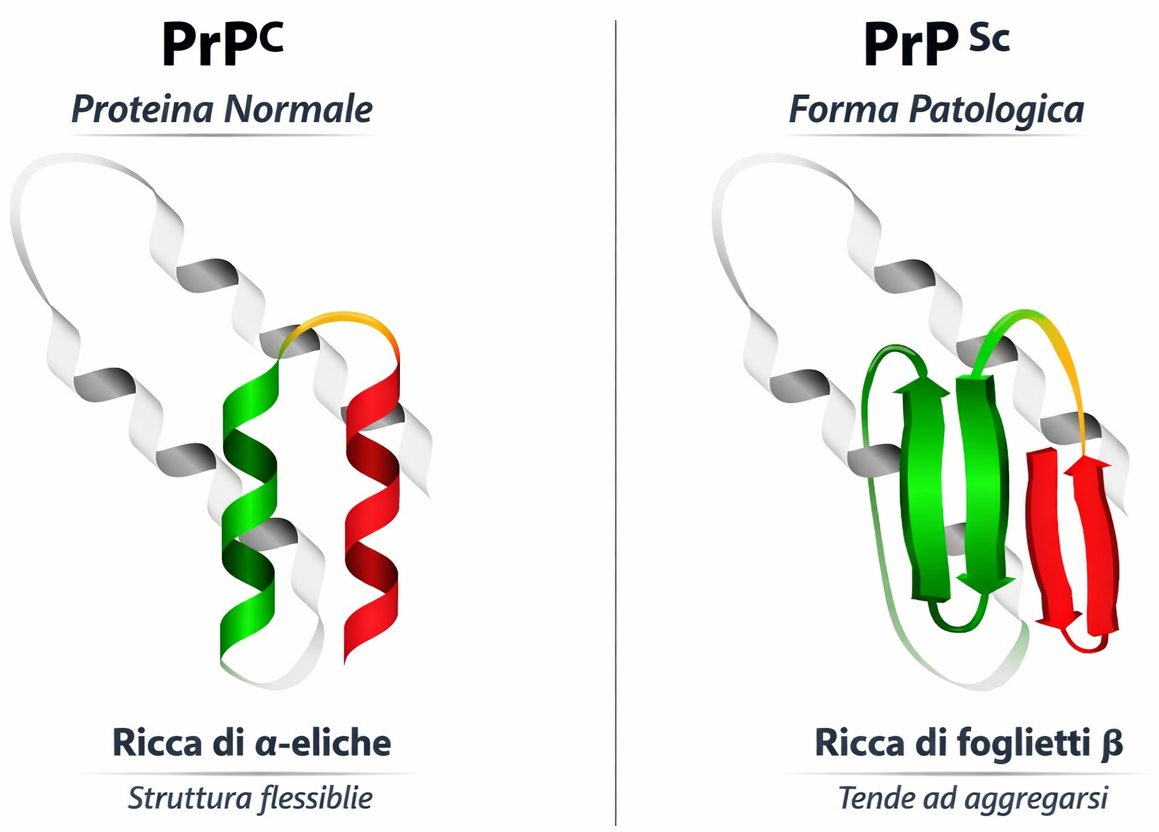
Un ripiegamento non corretto può indirizzare alcune proteine verso forme altamente ordinate note come fibrille amiloidi, strutture robuste con architettura a “cross-β” che tendono ad aggregare e a perturbare l’omeostasi cellulare. Questi assemblaggi sono implicati in numerose malattie neurodegenerative, tra cui il morbo di Alzheimer e la malattia di Huntington, nelle quali specie oligomeriche e fibrillari alterano funzioni sinaptiche, metabolismo proteico e vitalità neuronale. In un sottogruppo di encefalopatie spongiformi trasmissibili, l’agente eziologico è una proteina mal ripiegata, il prione, capace di indurre un rimodellamento conformazionale a stampo della corrispondente proteina nativa. Esempi classici includono la scrapie negli ovini, l’encefalopatia spongiforme bovina (BSE) nei bovini e la malattia di Creutzfeldt-Jakob (CJD) nell’uomo.
Nel sistema nervoso, la proteina prionica patologica converte la forma fisiologica nella conformazione anomala mediante un processo di “templated misfolding”. La conseguente nucleazione e polimerizzazione portano alla formazione di aggregati proteici (Figura 01.16-19), i quali possono diffondere lungo circuiti neuronali e tra cellule adiacenti. Tale propagazione, unitamente alla notevole resistenza alla proteolisi e ai trattamenti convenzionali di sterilizzazione, spiega la natura “infettiva” dei prioni e la loro trasmissione attraverso alimenti, sangue o strumenti chirurgici contaminati. Sebbene le cellule dispongano di sistemi di proteostasi (chaperonine, proteasoma, autofagia) per intercettare e degradare proteine anomale, un sovraccarico o un difetto di questi meccanismi può favorire l’accumulo amiloide e il danno tissutale.
Image Gallery
La struttura proteica si organizza su livelli gerarchici interdipendenti. La sequenza di amminoacidi determina la struttura primaria, dalla quale emergono gli elementi regolari di struttura secondaria, principalmente α-eliche e foglietti β, stabilizzati da legami a idrogeno lungo lo scheletro polipeptidico. L’insieme tridimensionale adottato dall’intera catena prende il nome di struttura terziaria e include, oltre a eliche e foglietti, anse, tratti irregolari e ripiegamenti che connettono l’estremità N-terminale a quella C-terminale. Quando più catene si associano in un complesso funzionale, la loro disposizione spaziale definisce la struttura quaternaria. Interazioni idrofobiche, legami a idrogeno, ponti disolfuro e contatti ionici cooperano nella stabilizzazione di questi livelli.
All’interno di molte proteine si riconoscono unità autonome, i domini, porzioni di 40–350 amminoacidi capaci di ripiegarsi indipendentemente in un nucleo compatto e stabile. I domini costituiscono i moduli funzionali e strutturali riutilizzati dall’evoluzione, spesso combinati in proteine di grandi dimensioni (Figura 01.16-20). Domini diversi possono mediare compiti distinti nella stessa molecola. Un esempio è la proteina batterica CAP (Figura 01.16-20), in cui un dominio lega il DNA, mentre un secondo dominio riconosce l’AMP ciclico. Il legame dell’AMP ciclico induce un’alterazione allosterica nel dominio di regolazione che abilita il dominio di legame al DNA a riconoscere una sequenza specifica, modulando così l’espressione genica.
Accanto ai domini, ricorrono motivi o “superstrutture” secondarie (per esempio, helix–turn–helix, β-hairpin) che fungono da mattoni ricorrenti in differenti contesti proteici. Modelli a nastro di domini di natura diversa, riportati nella (Figura 01.16-21), illustrano la varietà di questi moduli e le relazioni tra forma e funzione.
Image Gallery
Image Gallery
Image Gallery

Proteine di piccole dimensioni, come la mioglobina che trasporta ossigeno nel tessuto muscolare, tendono a essere costituite da un singolo dominio (Figura 01.16-10). Proteine più ampie possono integrare numerosi domini collegati da tratti di catena poveri di struttura regolare. Queste regioni intrinsecamente disordinate (IDR) non adottano un’unica conformazione stabile, ma fluttuano dinamicamente per via dell’agitazione termica.
Algoritmi bioinformatici, basati su proprietà come la bassa complessità di sequenza e la scarsità di residui idrofobici, hanno permesso di identificarle su larga scala. Si stima che circa un terzo delle proteine eucariotiche presenti segmenti disordinati più lunghi di 30 residui. L’assenza di struttura non è sinonimo di assenza di funzione: le IDR sono particolarmente adatte a interazioni transitorie e regolative. Tra i ruoli tipici si annoverano:
- segmenti flessibili di collegamento tra domini che consentono movimenti allosterici e rimodellamenti conformazionali;
- piattaforme per modificazioni post-traduzionali (per esempio fosforilazioni multiple) che integrano segnali cellula-specifici;
- motivi di riconoscimento a bassa affinità e alta specificità (SLiMs, MoRFs) per interazioni temporanee con partner diversi;
- partecipazione a condensati biomolecolari tramite separazione di fase, favorendo la compartimentazione senza membrana di processi come trascrizione e risposta allo stress.
Image Gallery
Con 20 amminoacidi, lo spazio delle sequenze cresce combinatoriamente: una catena di lunghezza \(n\) possiede \(20^n\) possibili disposizioni. Già per \(n=5\) si ottengono \(20^5 = 3,2\) milioni di sequenze; per una proteina di 200 residui, \(20^{200}\) supera di gran lunga \(10^{260}\). Solo una frazione esigua di questo spazio corrisponde a polipeptidi che si ripiegano in modo efficiente in una conformazione tridimensionale stabile, solubile e funzionalmente adeguata.
La selezione naturale ha favorito sequenze che massimizzano la stabilità del nucleo idrofobico, minimizzano la tendenza all’aggregazione e conferiscono le proprietà chimiche richieste dall’attività biologica. Piccole variazioni, anche limitate ad alcuni atomi in un residuo critico, possono perturbare il sito attivo, la dinamica conformazionale o l’interfaccia di legame, con perdita di funzione. Per contro, molti fold strutturali, e i domini che li incarnano, sono rimasti altamente conservati lungo linee evolutive distanti. Esempi notevoli includono domini di legame al DNA che mantengono la medesima topologia in regolatori trascrizionali di lieviti, animali e piante, nonostante oltre un miliardo di anni di divergenza.
L’equilibrio tra stabilità e funzione non è unico: esistono compromessi tra robustezza termodinamica, efficienza catalitica, specificità e prevenzione dell’aggregazione. La rete di proteostasi, con chaperoni e sistemi di degradazione, permette alle cellule di tollerare variazioni limitate, ma sequenze con forte propensione all’associazione indesiderata vengono generalmente eliminate durante l’evoluzione.
Una volta acquisiti un ripiegamento affidabile e proprietà vantaggiose, le proteine possono divergere per mutazione, duplicazione genica e riassortimento modulare, originando famiglie di omologhi con sequenze e strutture tridimensionali affini. Mantenendo una cornice strutturale condivisa, i membri della stessa famiglia possono specializzarsi in funzioni differenti attraverso modifiche locali di siti attivi, superfici di legame o elementi regolativi.
Le serina-proteasi rappresentano un caso paradigmatico. Questi enzimi proteolitici condividono un fold quasi sovrapponibile (Figura 01.16-22) e un medesimo nucleo catalitico, la triade Ser–His–Asp, ma differiscono per la specificità verso i substrati. Enzimi digestivi come tripsina e chimotripsina, e proteasi implicate nella coagulazione ematica quali trombina e fattore Xa, mostrano una forte conservazione dell’architettura globale; la discriminazione del legame peptidico da idrolizzare è determinata da cambiamenti mirati nelle tasche di specificità e nelle regioni di riconoscimento del substrato. L’elevata somiglianza delle catene, lunghe centinaia di residui, si traduce in tracciati tridimensionali pressoché identici, mentre modeste variazioni nelle aree funzionali producono attività distinte.
La classificazione in famiglie non si basa solo sulla sequenza, ma integra struttura e funzione. Omologhi ortologhi conservano in genere la funzione in specie diverse, mentre i paraloghi, derivati da duplicazioni all’interno dello stesso genoma, tendono a divergere funzionalmente. In molti casi, la ricombinazione di domini — per esempio, l’accoppiamento di un dominio catalitico con un dominio di legame a specifici partner — amplia il repertorio funzionale senza inventare ex novo interi ripiegamenti, sfruttando così la modularità intrinseca delle proteine.
Image Gallery
Nell’ambiente cellulare, le stesse forze deboli non covalenti che guidano il ripiegamento di una catena polipeptidica verso una conformazione definita promuovono anche l’associazione cooperativa tra proteine. La porzione di superficie che interagisce in modo complementare con un’altra molecola attraverso molteplici contatti (legami a idrogeno, interazioni ioniche, forze di van der Waals ed effetti idrofobici) costituisce un sito di legame. Una singola proteina può esibire molteplici siti con specificità distinte per ligandi sia piccoli sia macromolecolari. Quando la complementarità geometrica e chimica tra due superfici proteiche è elevata, l’associazione è stabile e definisce una struttura quaternaria caratteristica. In questo contesto, ciascuna catena polipeptidica di un complesso è detta subunità, e ogni subunità può essere a sua volta composta da più domini strutturali.
L’interazione tra subunità è descrivibile in termini di equilibrio di legame, ad esempio per un dimero omomero: \(P + P \rightleftharpoons P_2\), con \(K_d = \frac{[P]^2}{[P_2]}\). Valori di \(K_d\) nell’intervallo nanomolare–micromolare riflettono, rispettivamente, complessi molto stabili o più dinamici. Oltre a favorire la stabilità, l’oligomerizzazione introduce proprietà collettive non presenti nelle singole subunità, come allosteria e regolazione cooperativa:
- Vantaggi funzionali dell’oligomeria: aumento della stabilità termodinamica del complesso e riduzione della superficie esposta al solvente;
- modularità: combinazione di domini e subunità diverse per creare nuove funzioni catalitiche o regolatorie;
- regolazione: trasduzione di segnali allosterici tra siti di legame distanti e controllo della cooperatività;
- controllo della specificità: interfacce simmetriche limitano associazioni spurie e favoriscono architetture definite.
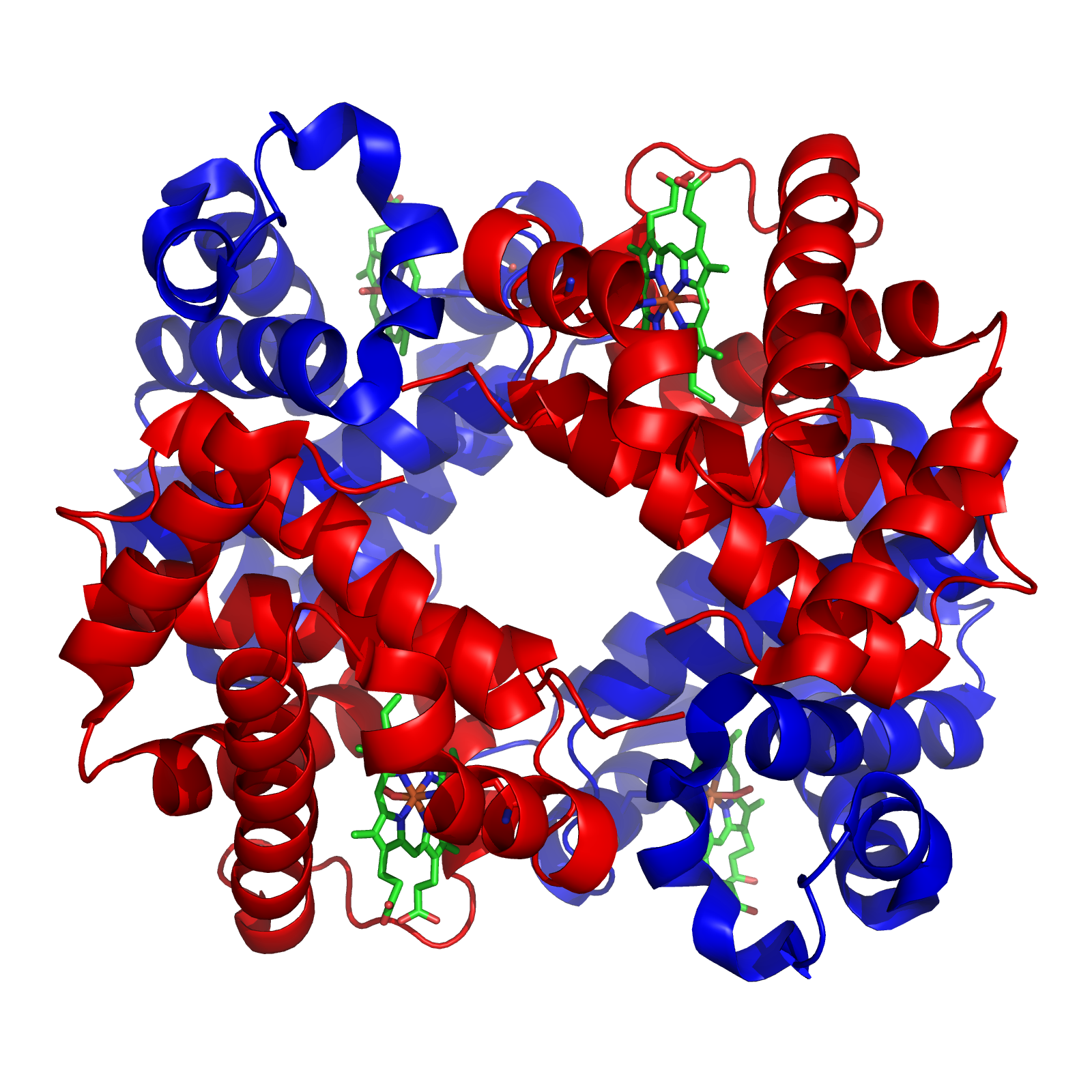
Nel caso più semplice, due catene identiche pre-ripiegate formano un dimero con simmetria semplice, mediato da un’interfaccia isologa tra siti di legame equivalenti. La proteina CAP dei batteri costituisce un esempio di omodimero (Figura 01.16-23) generato da due copie uguali della subunità illustrata in (Figura 01.16-20). Numerosi complessi cellulari mostrano simmetrie ripetute costruite a partire da una sola specie di subunità; la neuramminidasi è un tetramero con simmetria ciclica a quattro ripetizioni, organizzato in una struttura anulare (Figura 01.16-23). Accanto agli omooligomeri sono molto diffusi complessi eteromeri. L’emoglobina, la proteina trasportatrice di O2 degli eritrociti, è formata da due subunità di α-globina e due di β-globina, con disposizione simmetrica α2β2 (Figura 01.16-24); la sua architettura consente transizioni allosteriche tra stati conformazionali con diversa affinità per l’ossigeno.
Image Gallery

Image Gallery
Image Gallery
Image Gallery
Le proteine non si limitano a formare oligomeri compatti: spesso si auto-organizzano in strutture sovramolecolari estese. Un modo ricorrente è l’assemblaggio testa-coda lungo un asse, in cui un sito di legame su una molecola riconosce una regione complementare sulla successiva. Se l’interazione è ripetitiva, la catena di subunità adotta un’architettura elicoidale definita da un angolo di rotazione e da un avanzamento per subunità (Figura 01.16-14), consentendo una crescita potenzialmente illimitata in entrambe le direzioni (Figura 01.16-25). Un esempio emblematico è il filamento di actina, un polimero elicoidale allungato derivante dall’assemblaggio di molte unità di actina (Figura 01.16-26). Nell’eucariota, l’actina è fondamentale per la dinamica del citoscheletro e presenta polarità strutturale, con estremi “più” e “meno” che regolano la cinetica di crescita e disassemblaggio.
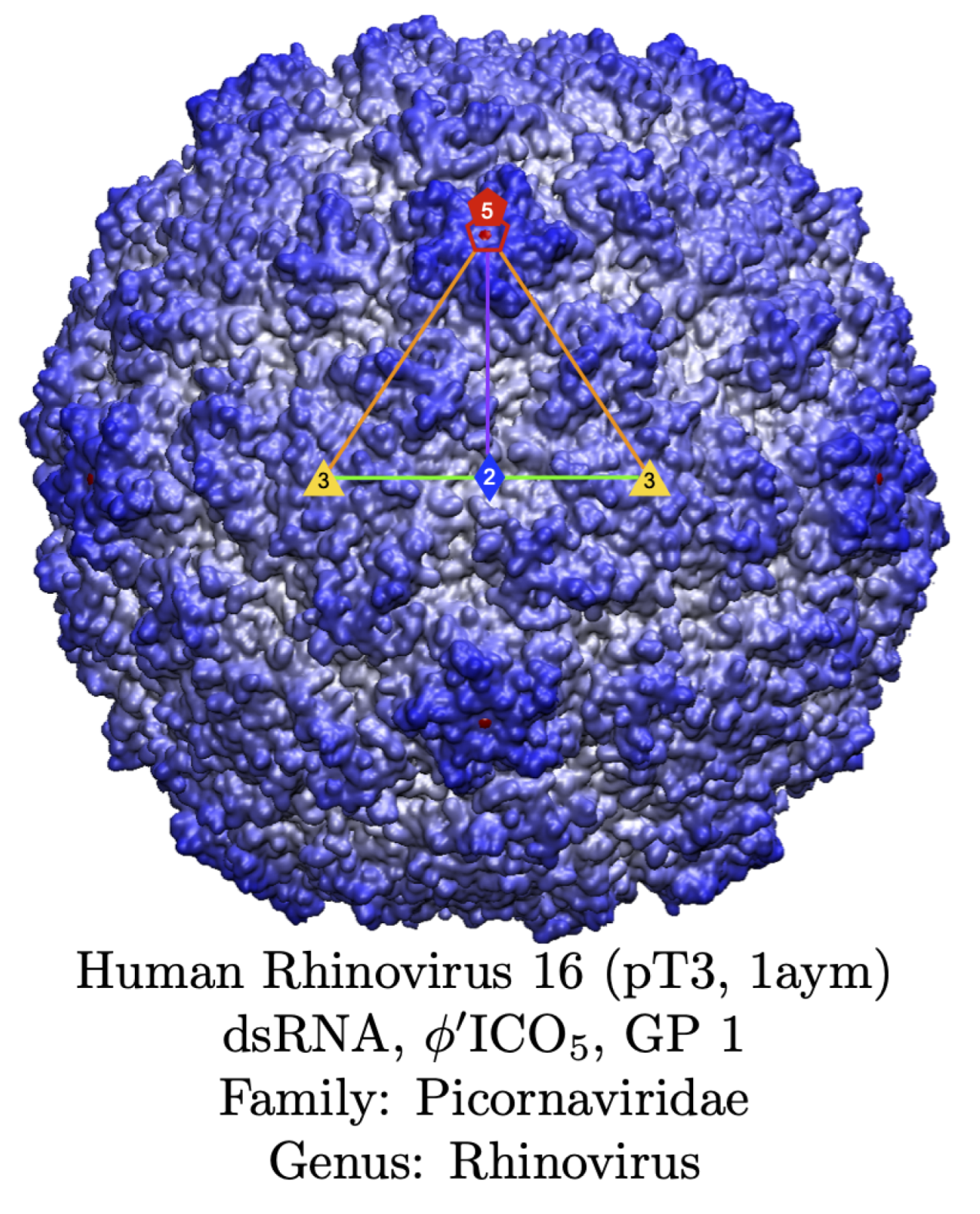
Organizzazioni alternative includono forme tubulari e involucri chiusi. I microtubuli (Figura 01.16-27) risultano dall’associazione laterale di protofilamenti di tubulina in un cilindro cavo con precisione geometrica; simmetricamente, molti virus incapsulano il proprio genoma in capside proteici che formano gusci sferici o icosaedrici (Figura 01.16-28), massimizzando stabilità con un numero limitato di tipi di subunità. Altre megastrutture, come ribosomi e ribonucleoproteine, combinano più specie proteiche con RNA o DNA in architetture a funzione altamente coordinata. Questi complessi possono essere isolati, dissociati nei costituenti e, in condizioni adeguate di composizione e ambiente, riassemblati spontaneamente. Tale comportamento indica che l’informazione necessaria all’autoassemblaggio è codificata nelle superfici delle macromolecole; la formazione è termodinamicamente favorita quando \( \Delta G = \Delta H - T\Delta S < 0 \), condizione che dipende da complementarità strutturale, concentrazione e ioni cofattori.
Image Gallery
Image Gallery
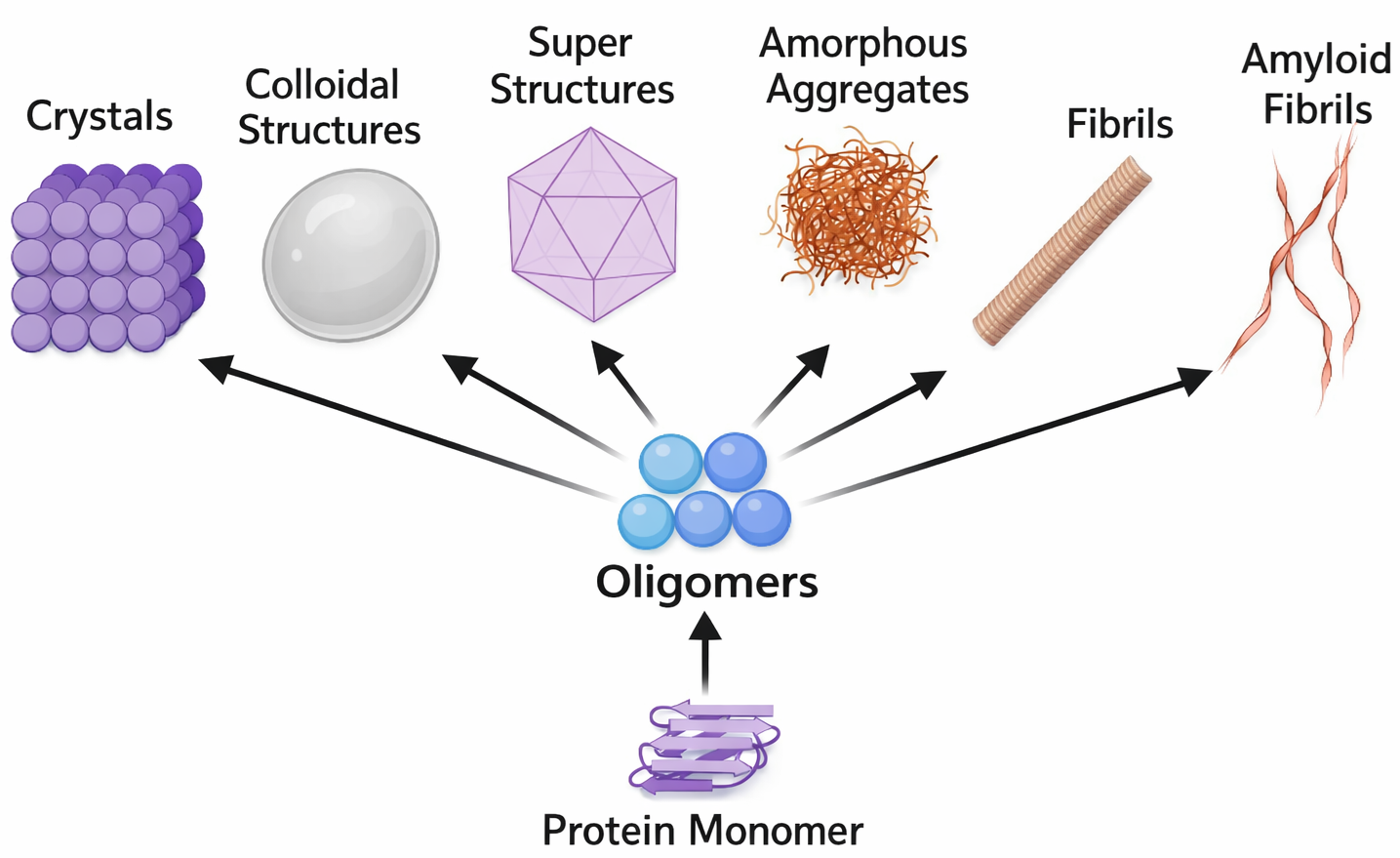
Image Gallery
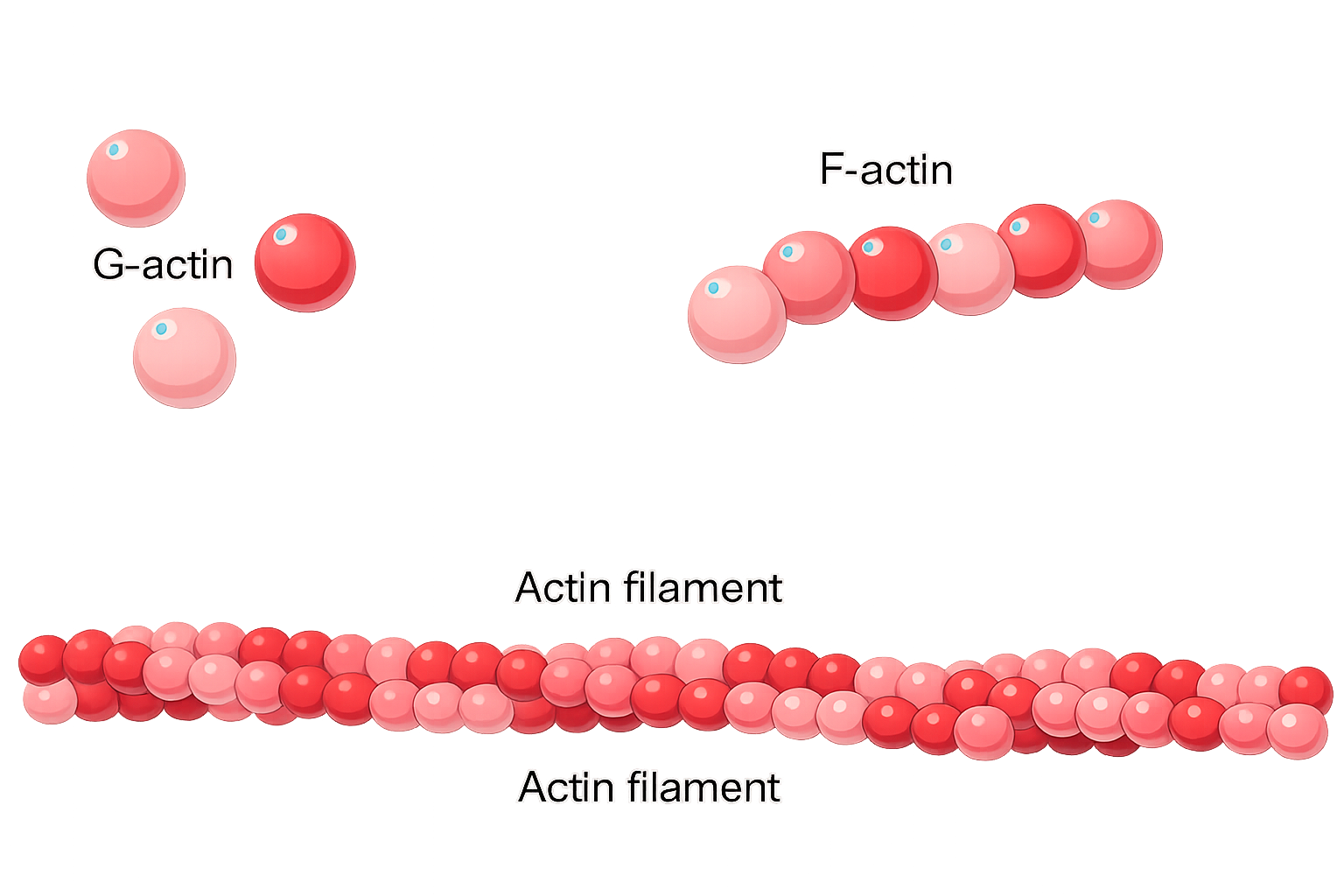
Image Gallery
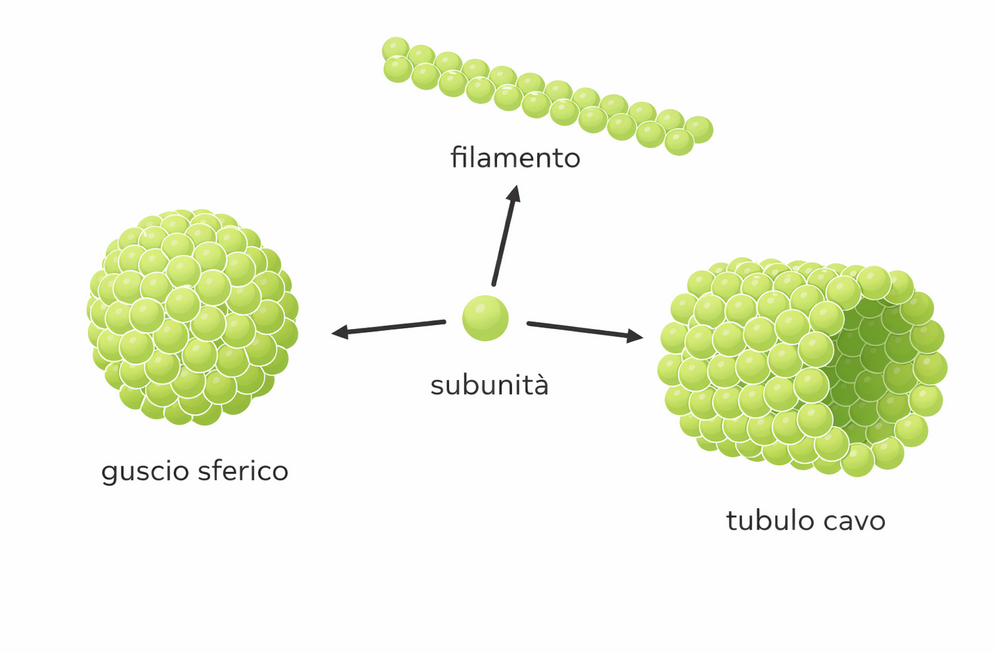
Image Gallery
Molti enzimi e regolatori intracellulari sono proteine globulari, con catene polipeptidiche ripiegate in agglomerati compatti e superfici irregolari adatte al riconoscimento molecolare (Figura 01.16-10). In altri contesti funzionali è invece richiesto che la singola molecola copra distanze maggiori o trasferisca forze meccaniche: in tali casi prevalgono architetture allungate relativamente semplici, le cosiddette proteine fibrose.
Una famiglia ampia di proteine fibrose intracellulari è rappresentata dalle cheratine di tipo α, già incontrate in relazione all’α-elica. Le loro unità fondamentali sono dimeri a coiled-coil, in cui due α-eliche parallele si avvolgono l’una sull’altra secondo un registro eptadico caratteristico, stabilizzato da residui idrofobici nei siti a e d e da specifiche interazioni laterali (Figura 01.16-16). Le estremità delle regioni coiled-coil ospitano domini globulari con siti di interazione che mediano l’assemblaggio gerarchico in filamenti intermedi, elementi del citoscheletro che impartiscono resistenza meccanica a cellule e tessuti. L’elevata stabilità dei filamenti di cheratina spiega la persistenza di strutture come capelli, unghie e rivestimenti cornei.
All’esterno della cellula, proteine fibrose costituiscono l’impalcatura della matrice extracellulare, una rete idratata che conferisce coesione ai tessuti. Il collagene è la componente più abbondante nei vertebrati. Ogni molecola di procollagene comprende tre catene polipeptidiche che, grazie a una ripetizione quasi regolare Gly–X–Y (con X e Y spesso Pro e idrossi-Pro), si avvolgono in una tripla elica con la glicina rivolta verso l’asse centrale, dove è essenziale il suo raggio sterico minimo (Figura 01.16-29). Più tripliche eliche si allineano in modo sfalsato testa-coda e fianco a fianco dando origine a fibrille di collagene estremamente robuste, la cui resistenza deriva sia dall’impaccamento ordinato sia da legami crociati covalenti maturati enzimaticamente su residui di lisina. La corretta biosintesi del collagene dipende da modificazioni post-traduzionali, come l’idrossilazione di Pro e Lys, che stabilizzano la tripla elica mediante legami a idrogeno addizionali.
In netto contrasto strutturale con il collagene, l’elastina forma reti flessibili. Le sue catene sono ricche di segmenti idrofobici disordinati e vengono interconnesse da legami covalenti specifici (ad esempio, desmosina e isodesmosina derivanti da lisina), generando maglie elastomeriche a comportamento “gommoso”. Le fibre elastiche permettono a pelle, vasi arteriosi e parenchima polmonare di allungarsi e recuperare la lunghezza originaria senza frattura; l’energia elastica ha natura prevalentemente entropica, poiché il tratto “stirato” riduce la varietà conformazionale disponibile, mentre il rilascio ripristina lo stato a maggiore entropia (Figura 01.16-29).
Image Gallery
Image Gallery
Image Gallery
Image Gallery
Proteine ancorate alla superficie cellulare o secrete nella matrice extracellulare incontrano condizioni più ossidanti, variazioni di pH e stress meccanici che richiedono stabilizzazioni aggiuntive. Un meccanismo diffuso è l’introduzione di legami covalenti crociati, che possono collegare due residui all’interno della stessa catena (intramolecolari) o unire catene distinte (intermolecolari), come avviene nelle fibrille collageniche o nelle reti elastiche. Tali crociamenti limitano fluttuazioni indotte dal solvente e fissano configurazioni favorevoli dal punto di vista energetico.
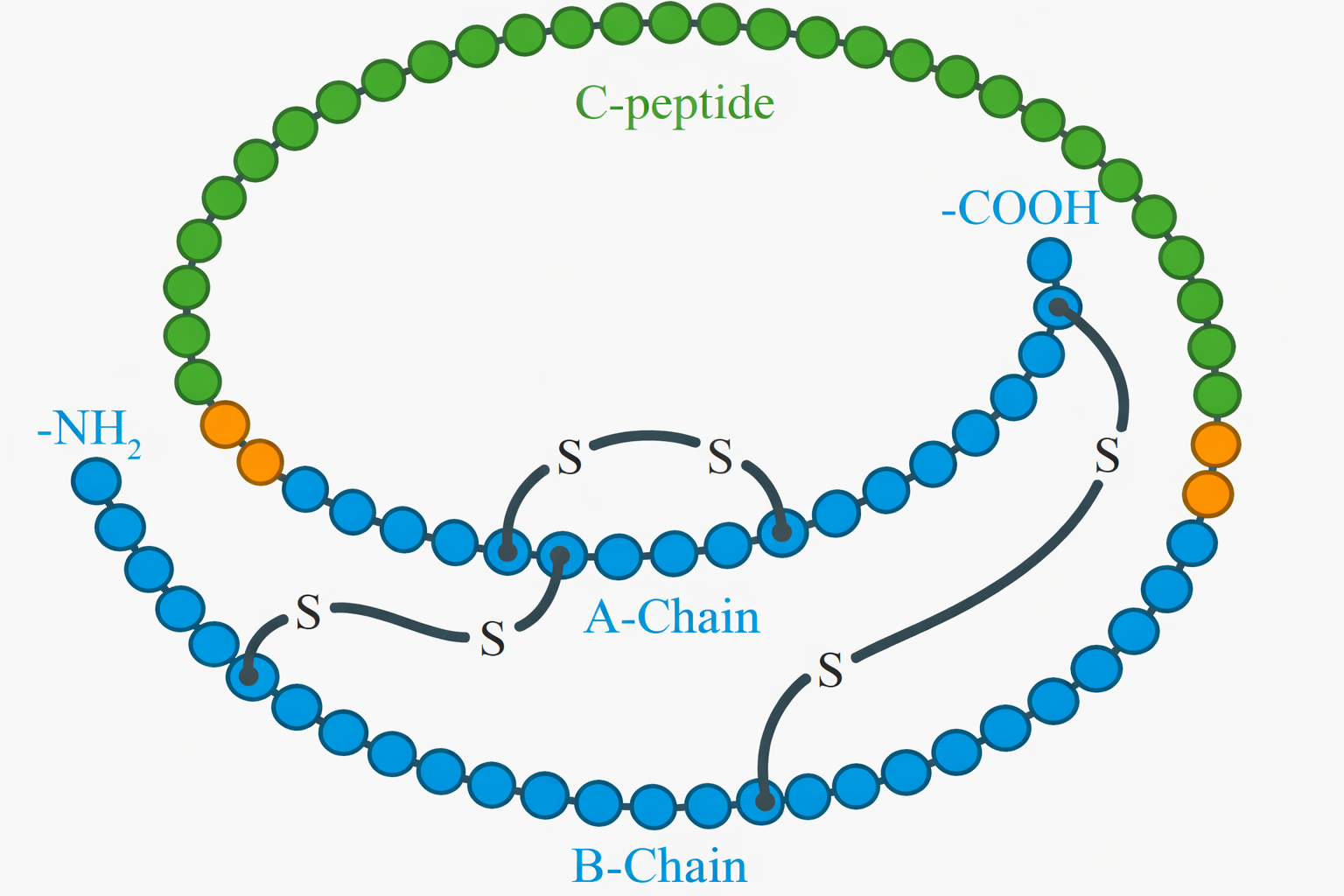
I legami crociati più comuni sono i ponti disolfuro, ossia legami S–S tra due cisteine. Nella via secretoria, il reticolo endoplasmatico fornisce un ambiente ossidante e gli enzimi proteina-disolfuro isomerasi catalizzano sia la formazione sia il riarrangiamento dei ponti per ottenere l’assetto nativo (Figura 01.16-30). I ponti disolfuro non impongono nuove pieghe, ma agiscono come “graffette atomiche” che consolidano la conformazione più stabile. Un esempio classico è il lisozima, presente in lacrime e saliva, la cui attività antibatterica persiste a lungo grazie a un reticolo di ponti disolfuro che ne riduce la suscettibilità alla denaturazione. Anche le immunoglobuline sfruttano disolfuri per connettere domini e catene, conferendo tenuta meccanica e resistenza proteasica.
In citosol, la formazione di S–S è sfavorita dall’ambiente fortemente riducente (rapporto elevato GSH/GSSG), per cui i gruppi tiolici della cisteina rimangono prevalentemente in forma –SH. Non sorprende quindi che le proteine citosoliche non necessitino di tali rinforzi, mentre compartimenti più ossidanti, come lume del reticolo ed endomembrane o il periplasma batterico, promuovono la maturazione di ponti disolfuro. Oltre ai disolfuri, esistono altri legami crociati rilevanti: tra essi, i ponti catalizzati da lisil ossidasi nelle fibrille di collagene e i legami ε-(γ-glutamil)–lisina mediati da transglutaminasi, che aumentano la resistenza dei tessuti;
in sintesi, l’equilibrio tra autoassemblaggio governato da interazioni non covalenti e “cuciture” covalenti consente alle proteine di costruire complessi modulari, dinamici e al contempo sufficientemente robusti per operare in contesti cellulari ed extracellulari.
Image Gallery