


Pochi gruppi chimici sono fondamentali per le funzioni molecolari
TOPICS
Definizione

Nelle proteine, organizzazione tridimensionale e funzione biologica sono inscindibili. La disposizione spaziale delle catene laterali e la loro chimica di superficie, entrambe altamente specifiche, determinano la capacità della proteina di riconoscere partner molecolari e di svolgere compiti distinti. Dall’integrazione tra architettura, reattività e dinamica scaturisce la versatilità delle proteine nel coordinare i processi cellulari. La domanda centrale è: in virtù di quali principi le proteine esercitano le loro funzioni? Il filo conduttore è il legame selettivo a molecole complementari, che consente alle proteine di agire da catalizzatori, elementi strutturali, motori, sensori e regolatori. Gli esempi discussi rappresentano un sottoinsieme dell’ampia gamma di funzioni, ma poggiano su meccanismi comuni e ripetibili.
Le proprietà biologiche di una proteina emergono dalle sue interazioni fisiche con altre molecole. Anticorpi che si attaccano a virus o batteri per neutralizzarli, esochinasi che lega D-glucosio e ATP per trasferire un fosfato, subunità di actina che si associano in filamenti: in tutti i casi il legame è specifico e può avere affinità molto diverse, dalla transitoria all’eccezionalmente stabile.
La molecola che si associa a una proteina è detta ligando. La specificità di riconoscimento nasce dalla complementarità tra superfici: il sito di legame proteico presenta una configurazione geometrica e chimica in grado di instaurare un gran numero di interazioni non covalenti con il ligando, tra cui legami idrogeno, interazioni elettrostatiche e forze di van der Waals, alle quali si sommano contributi idrofobici. Ciascuna interazione, considerata singolarmente, è debole; la somma cooperativa di molte interazioni, resa possibile dalla complementarità dei profili molecolari, determina l’elevata affinità complessiva, secondo un principio spesso rappresentato come “guanto e mano” (Figura 01.17-01).
Quando la complementarità è scarsa, si instaurano poche interazioni e il complesso si dissocia rapidamente, prevenendo associazioni aspecifiche. Al contrario, un’ampia rete di contatti non covalenti produce associazioni persistenti, essenziali quando la funzione lo richiede, come avviene negli assemblaggi macromolecolari stabili, ad esempio il ribosoma.
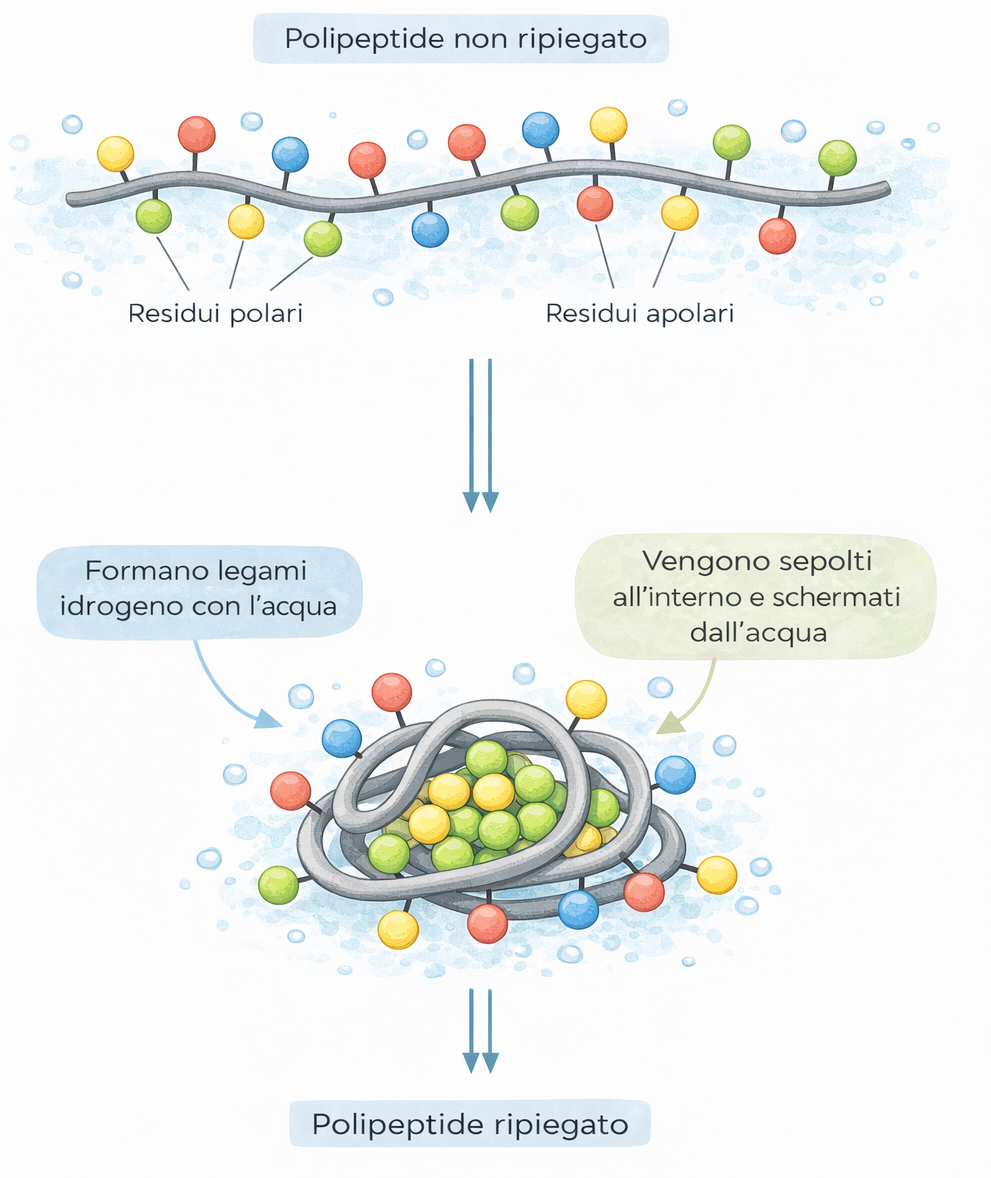
Il sito di legame, in genere una cavità o un solco sulla superficie, è costruito dalla disposizione di catene laterali di amminoacidi che, pur lontane nella sequenza lineare, vengono a contatto durante il ripiegamento (Figura 01.17-02). Spesso una proteina ospita più siti: alcuni dedicati a ligandi regolatori che modulano l’attività, altri a segnali per il posizionamento subcellulare. Nelle proteine di membrana, segmenti idrofobi ad α-elica facilitano l’inserzione e la stabilizzazione nel doppio strato lipidico (Figura 01.17-10).
Gli atomi interni della proteina, anche se non toccano direttamente il ligando, sostengono l’architettura del sito di legame e ne definiscono la chimica di superficie. Piccole sostituzioni di residui nel nucleo idrofobo possono alterare la conformazione globale e compromettere affinità e selettività.
La termodinamica e la cinetica del legame forniscono una descrizione quantitativa utile. L’affinità è misurata dalla costante di dissociazione \(K_d\), con valori tipici che spaziano da picomolare ad alta affinità a millimolare per legami deboli; a parità di condizioni, la frazione di occupazione del sito è \( \theta = \frac{[L]}{K_d + [L]} \). La velocità di associazione e dissociazione è descritta da \(k_{on}\) e \(k_{off}\), con \(K_d = \frac{k_{off}}{k_{on}}\). Molte proteine mostrano adattamento conformazionale all’atto del legame (induced fit) o selezionano, tra conformazioni preesistenti, quella più compatibile (conformational selection), migliorando ulteriormente la complementarità:
- determinanti della specificità: complementarità di forma e carica, distribuzione di donatori/accettori di H, topologia idrofobica e polarità;
- stabilità del complesso: somma cooperativa di interazioni deboli e contributo del solvente;
- controllo dell’affinità: microambiente locale, flessibilità conformazionale, presenza di cofattori o ioni;
- conseguenze funzionali: durata del complesso in relazione al compito da svolgere (transitorio per segnalazione, stabile per strutture assemblate).
Image Gallery
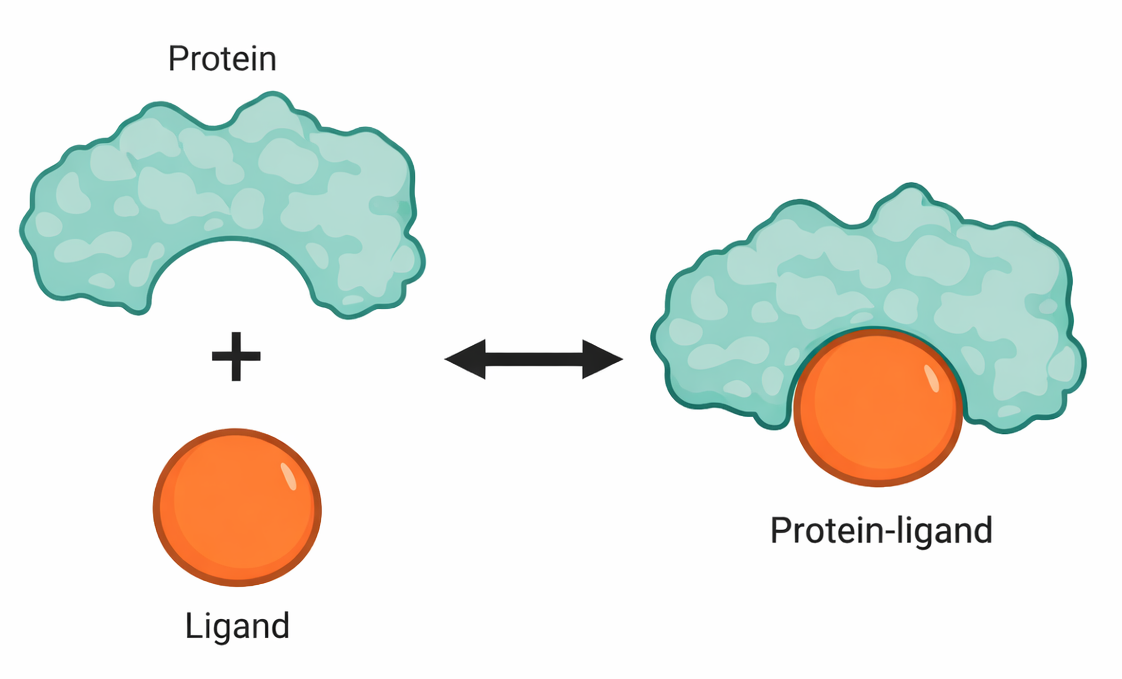
Image Gallery
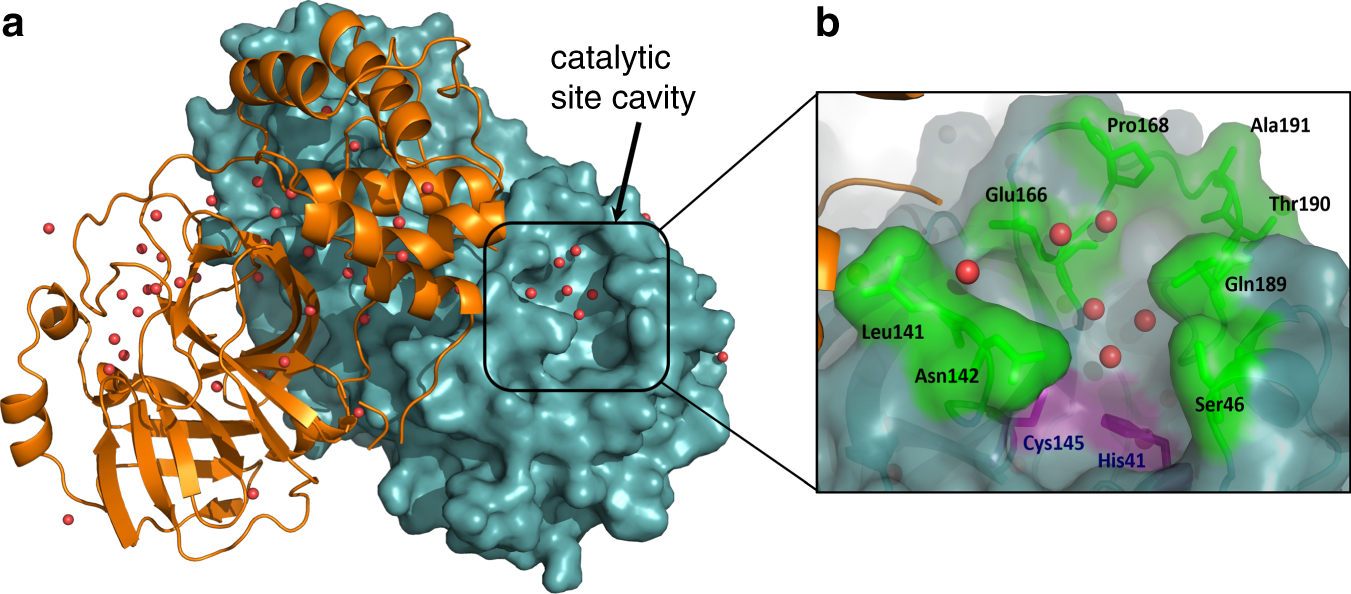
Image Gallery
Tutte le proteine richiedono legami specifici per operare; gli anticorpi spiccano per l’ampiezza dello spettro di ligandi riconoscibili, comprendente componenti di batteri, virus e altri patogeni. Le immunoglobuline sono prodotte in risposta a molecole estranee e si legano con grande affinità ai relativi bersagli, chiamati antigeni, inibendone l’azione o promuovendone l’eliminazione.
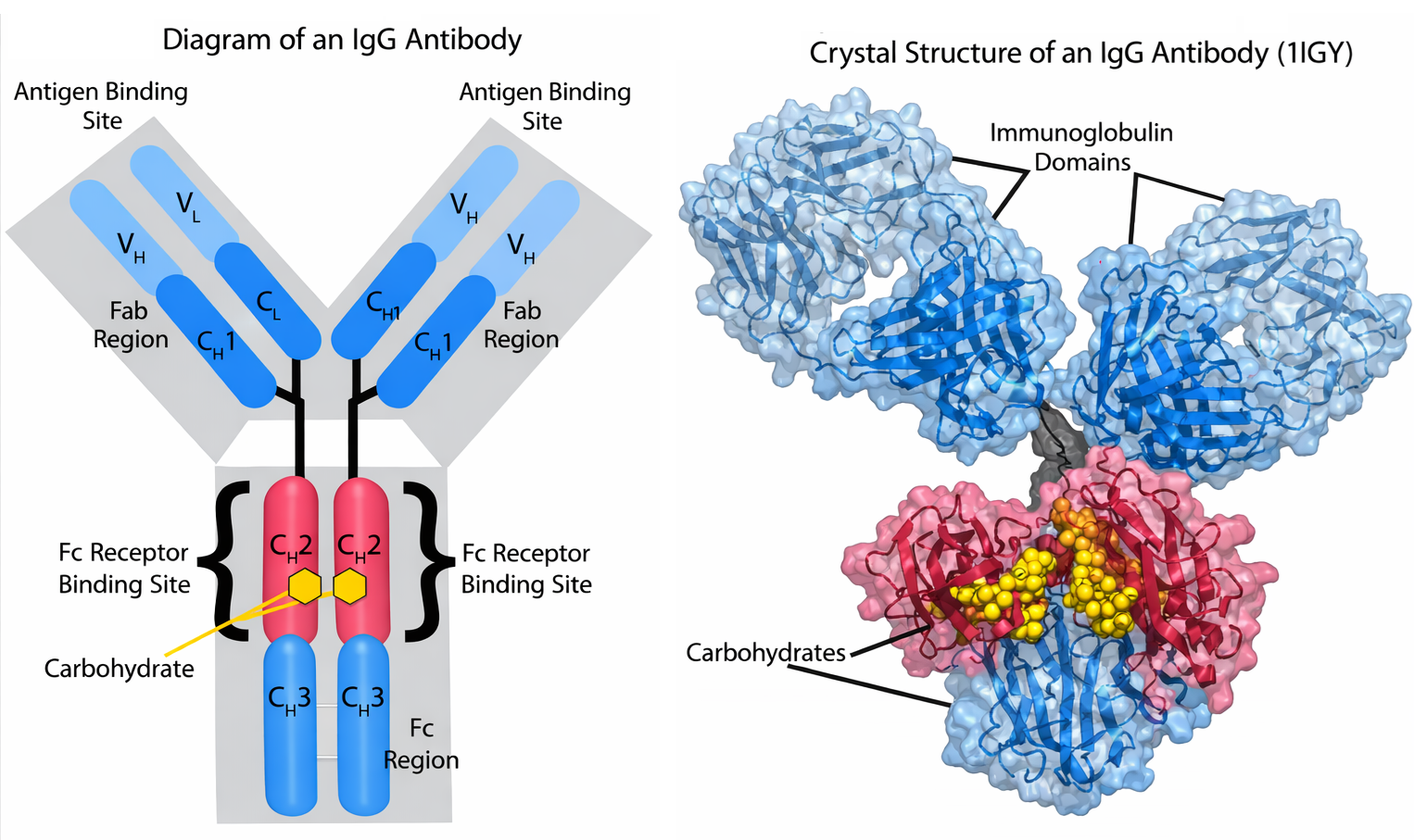
La straordinaria diversità degli anticorpi permette di coprire un repertorio potenzialmente pari a miliardi di antigeni. Ogni anticorpo presenta una struttura a Y con due siti di legame identici, ciascuno complementare a una porzione limitata della superficie antigenica. I siti appartengono a regioni variabili costituite da anse di catena polipeptidica che sporgono alle estremità di due domini adiacenti (Figura 01.17-03). La variazione in lunghezza e sequenza di queste anse, mantenendo intatto l’impalcato strutturale, genera una gamma vastissima di superfici di riconoscimento.
A livello molecolare, la specificità deriva dalla combinazione di contatti multipli tra residui variabili dell’anticorpo e epitopi antigenici. Nei linfociti B, la ricombinazione somatica dei segmenti genici e la mutazione somatica, seguite da selezione per affinità crescente, affinano nel tempo la complementarità, incrementando l’affinità fino a ordini di grandezza inferiori nel \(K_d\). Questa plasticità rende gli anticorpi strumenti irrinunciabili sia nella difesa immunitaria sia in ambito sperimentale per identificare, isolare e quantificare molecole specifiche.
Image Gallery
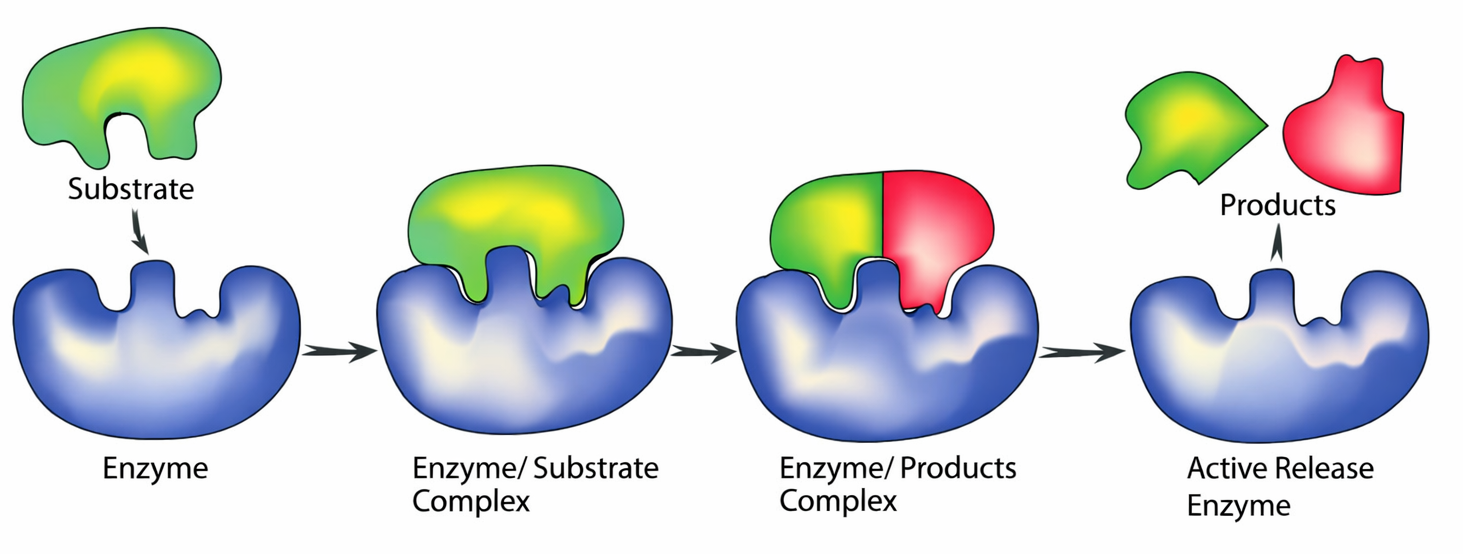
Alcune proteine hanno come compito esclusivo l’assemblaggio con copie di se stesse o con partner strutturali, come avviene per l’actina. In molte altre, il legame al ligando è il primo passo della catalisi chimica: questi sono gli enzimi. Gli enzimi si associano a uno o più substrati e li trasformano in prodotti modificati, restando invariati al termine del ciclo catalitico (Figura 01.17-04). In tal modo permettono la formazione e rottura di legami covalenti a velocità compatibili con la vita, orchestrando reti di reazioni che generano energia e costruiscono i componenti cellulari.
Ogni enzima è finemente selettivo per tipo di reazione e per substrato. L’esochinasi catalizza la fosforilazione del D-glucosio, ma non del suo enantiomero L; la trombina riconosce e cliva un sito peptidico ben definito, tra arginina e glicina. Le reazioni enzimatiche sono organizzate in vie metaboliche, in cui il prodotto di un enzima diventa substrato del successivo, consentendo un controllo modulare e la distribuzione efficiente dei flussi metabolici (Tabella 01.17-01).
La catalisi si fonda sulla stabilizzazione dello stato di transizione e sulla riduzione della barriera di attivazione. Tra le strategie ricorrenti figurano catalisi acido-base, catalisi covalente, coordinazione metallica e posizionamento ideale dei gruppi reattivi. La cinetica di Michaelis–Menten descrive molte reazioni a substrato singolo, con parametri \(K_m\) (affinità apparente) e \(V_{max}\) (velocità massima). A titolo illustrativo, un enzima con \(K_m = 50\,\mu\)M e \(k_{cat} = 100\,s^{-1}\) converte, a saturazione, ciascun sito attivo in 100 molecole di prodotto al secondo; l’efficienza catalitica \(k_{cat}/K_m\) confronta la prestazione tra enzimi e può avvicinarsi al limite di diffusione per catalizzatori particolarmente efficienti:
- determinanti della specificità: complementarità del sito attivo con il substrato, esclusione sterica di isomeri o gruppi non desiderati, reti di legami idrogeno direzionali;
- contributi meccanicistici: acido-base generale, residui nucleofili per intermedi covalenti, metalli per polarizzazione dei legami e stabilizzazione di cariche;
- regolazione: allosteria mediata da ligandi distanti dal sito attivo, modificazioni post-traduzionali, compartimentalizzazione;
- integrazione in reti: multienzimi e canali di trasferimento del substrato riducono la dissipazione e aumentano l’efficienza.
La classificazione funzionale degli enzimi si basa sul tipo di trasformazione chimica catalizzata e consente di sistematizzare la varietà delle reazioni cellulari (Tabella 01.17-01). Pur nella diversità dei meccanismi, rimangono invariati i principi cardine già menzionati: riconoscimento selettivo dei substrati, complementarità chimico-geometrica e cooperazione di molte interazioni deboli nel sito attivo, integrate da aggiustamenti conformazionali che ottimizzano la catalisi.
Image Gallery
Classi comuni di enzimi e loro funzione
| Classe enzimatica | Funzione biochimica |
|---|---|
| Idrolasi | Catalizzano scissioni idrolitiche in generale |
| Nucleasi | Degradano acidi nucleici rompendo i legami tra nucleotidi |
| Proteasi | Scindono proteine idrolizzando i legami peptidici |
| Ligasi | Uniscono due molecole, come la DNA ligasi che collega frammenti di DNA |
| Isomerasi | Riorganizzano legami all’interno di una stessa molecola |
| Polimerasi | Catalizzano la sintesi di polimeri come DNA e RNA |
| Chinasi | Aggiungono gruppi fosfato a molecole bersaglio, soprattutto proteine |
| Fosfatasi | Rimuovono gruppi fosfato da substrati molecolari |
| Ossidoreduttasi | Catalizzano reazioni di ossidazione o riduzione |
| ATPasi | Idrolizzano ATP fornendo energia a processi cellulari come trasporto e movimento |
| Tabella che riassume la classificazione degli enzimi in base al tipo di reazione catalizzata: scissione di legami, formazione di legami, trasferimento di gruppi funzionali o di elettroni. Questa suddivisione evidenzia il ruolo fondamentale degli enzimi nella regolazione delle vie metaboliche e nei principali processi cellulari. | |
Classi comuni di enzimi e loro funzione
| Classe enzimatica | Funzione biochimica |
|---|---|
| Idrolasi | Catalizzano scissioni idrolitiche in generale |
| Nucleasi | Degradano acidi nucleici rompendo i legami tra nucleotidi |
| Proteasi | Scindono proteine idrolizzando i legami peptidici |
| Ligasi | Uniscono due molecole, come la DNA ligasi che collega frammenti di DNA |
| Isomerasi | Riorganizzano legami all’interno di una stessa molecola |
| Polimerasi | Catalizzano la sintesi di polimeri come DNA e RNA |
| Chinasi | Aggiungono gruppi fosfato a molecole bersaglio, soprattutto proteine |
| Fosfatasi | Rimuovono gruppi fosfato da substrati molecolari |
| Ossidoreduttasi | Catalizzano reazioni di ossidazione o riduzione |
| ATPasi | Idrolizzano ATP fornendo energia a processi cellulari come trasporto e movimento |
| Tabella che riassume la classificazione degli enzimi in base al tipo di reazione catalizzata: scissione di legami, formazione di legami, trasferimento di gruppi funzionali o di elettroni. Questa suddivisione evidenzia il ruolo fondamentale degli enzimi nella regolazione delle vie metaboliche e nei principali processi cellulari. | |
L’affinità di un enzima per il suo substrato e la rapidità con cui il complesso enzima–substrato genera prodotto differiscono ampiamente tra enzimi diversi e possono essere quantificate in vitro utilizzando preparazioni purificate. Quando la concentrazione di substrato è bassa, la velocità iniziale di reazione è proporzionale a [S], perché la formazione del complesso enzima–substrato è l’evento che limita il processo. All’aumentare di [S], si raggiunge una condizione in cui quasi tutte le molecole enzimatiche risultano occupate: la velocità diviene allora insensibile a ulteriori incrementi di [S] e tende a un valore limite, la velocità massima \(V_{\max}\), determinata dal ritmo intrinseco con cui avviene la trasformazione del substrato in prodotto.
Per un’ampia classe di enzimi a cinetica di Michaelis–Menten, la dipendenza della velocità iniziale dalla concentrazione di substrato è descritta da:
\[ v = \frac{V_{\max}[S]}{K_M + [S]} \]
Il parametro \(K_M\) è la concentrazione di substrato alla quale \(v = \tfrac{1}{2}V_{\max}\) (Figura 01.17-05). La grandezza \(k_{\text{cat}}\) (numero di turnover) esprime quante molecole di substrato sono convertite in prodotto per unità di tempo da una singola molecola di enzima alla saturazione di substrato, con \(k_{\text{cat}} = \tfrac{V_{\max}}{[E]_T}\). Per molti enzimi \(k_{\text{cat}}\) è dell’ordine di 10^2–10^4 s\(^{-1}\), ma sono noti casi da pochi eventi al secondo fino a 10^5–10^6 s\(^{-1}\). In termini relativi, l’azione catalitica di un enzima può incrementare la velocità di reazione di 10^6 volte o più rispetto alla reazione non catalizzata.
Dal punto di vista interpretativo:
- un \(K_M\) piccolo segnala un’interazione molto stabile tra enzima e substrato, favorita da numerose interazioni non covalenti complementari nel sito attivo;
- un \(K_M\) elevato suggerisce un legame debole e una minore occupazione del sito attivo alle stesse concentrazioni di substrato;
- il rapporto \(k_{\text{cat}}/K_M\) misura l’efficienza catalitica alle basse [S] e confronta in modo utile enzimi diversi o varianti dello stesso enzima.
Esempio numerico: se un esperimento fornisce \(V_{\max} = 2\,\mu\text{M}\,\text{s}^{-1}\) e la concentrazione totale di enzima è \([E]_T = 10\,\text{nM}\), allora \(k_{\text{cat}} = 200\,\text{s}^{-1}\). Se \(K_M = 5\,\mu\text{M}\), l’efficienza catalitica risulta \(k_{\text{cat}}/K_M = 40\,\text{s}^{-1}\,\mu\text{M}^{-1}\).
Image Gallery

Per chiarire come un enzima realizzi la conversione chimica del substrato, è istruttivo considerare il lisozima, una piccola proteina presente nelle lacrime, nella saliva, nell’albume e in altre secrezioni, dove svolge una funzione antimicrobica. Il lisozima idrolizza i legami glicosidici β(1→4) delle catene polisaccaridiche del peptidoglicano nella parete batterica; la rottura anche di un numero limitato di tali legami indebolisce la parete fino a causare la lisi cellulare per effetto della pressione osmotica. La struttura atomica del lisozima, tra le prime determinate mediante cristallografia a raggi X, ha reso possibile correlare in modo fine struttura e funzione.
La reazione catalizzata è un’idrolisi: l’inserzione di una molecola d’acqua nel legame tra due zuccheri adiacenti scinde la catena, con una diminuzione dell’energia libera complessiva del sistema. Tuttavia, la reazione non procede in modo apprezzabile in assenza di catalisi perché la barriera energetica, l’energia di attivazione, è elevata; per rompere il legame glicosidico, il substrato deve acquisire una conformazione e una distribuzione elettronica proprie dello stato di transizione.
Il lisozima, come altri enzimi, possiede un sito attivo, una tasca o solco in cui il substrato si lega con geometria complementare e tramite molteplici interazioni deboli (Figura 01.17-02). Nel caso del lisozima il solco vincola simultaneamente sei unità saccaridiche della catena; il legame induce una distorsione in una specifica unità zuccherina, portandola in una conformazione “sforzata” che somiglia allo stato di transizione. In tale microambiente, gruppi catalitici opportunamente posizionati abbassano l’energia di attivazione (Figura 01.17-07) e favoriscono l’attacco nucleofilo dell’acqua al legame da scindere.
Gli studi strutturali hanno evidenziato un ruolo centrale di due residui acidi conservati: uno funge da acido generale protonando l’ossigeno glicosidico, l’altro stabilizza il carattere cationico del centro reattivo e, in alcuni modelli, forma un intermedio covalente con il substrato. Dopo le tappe successive di trasferimento protonico e attacco dell’acqua, il legame è rotto, i frammenti vengono rilasciati e l’enzima torna nello stato iniziale pronto per un nuovo ciclo (Figura 01.17-06). Grazie a questa combinazione di legame specifico, distorsione del substrato e catalisi acido–base (ed eventualmente covalente), la velocità globale risulta milioni di volte superiore rispetto alla reazione in soluzione acquosa priva di enzima.
Strategie analoghe sono impiegate diffusamente da altri enzimi per ridurre l’energia di attivazione: il sito attivo può allineare più substrati per favorire la reazione (Figura 01.17-08), modulare la distribuzione elettronica mediante gruppi polari opportunamente orientati (Figura 01.17-08) e imporre tensioni conformazionali che stabilizzano lo stato di transizione (Figura 01.17-08). In diversi casi, la formazione transitoria di legami covalenti tra una catena laterale del sito attivo e il substrato contribuisce alla catalisi; tali legami vengono poi idrolizzati o riorganizzati ripristinando l’enzima alla fine del ciclo.
Image Gallery
Image Gallery

Image Gallery

Image Gallery
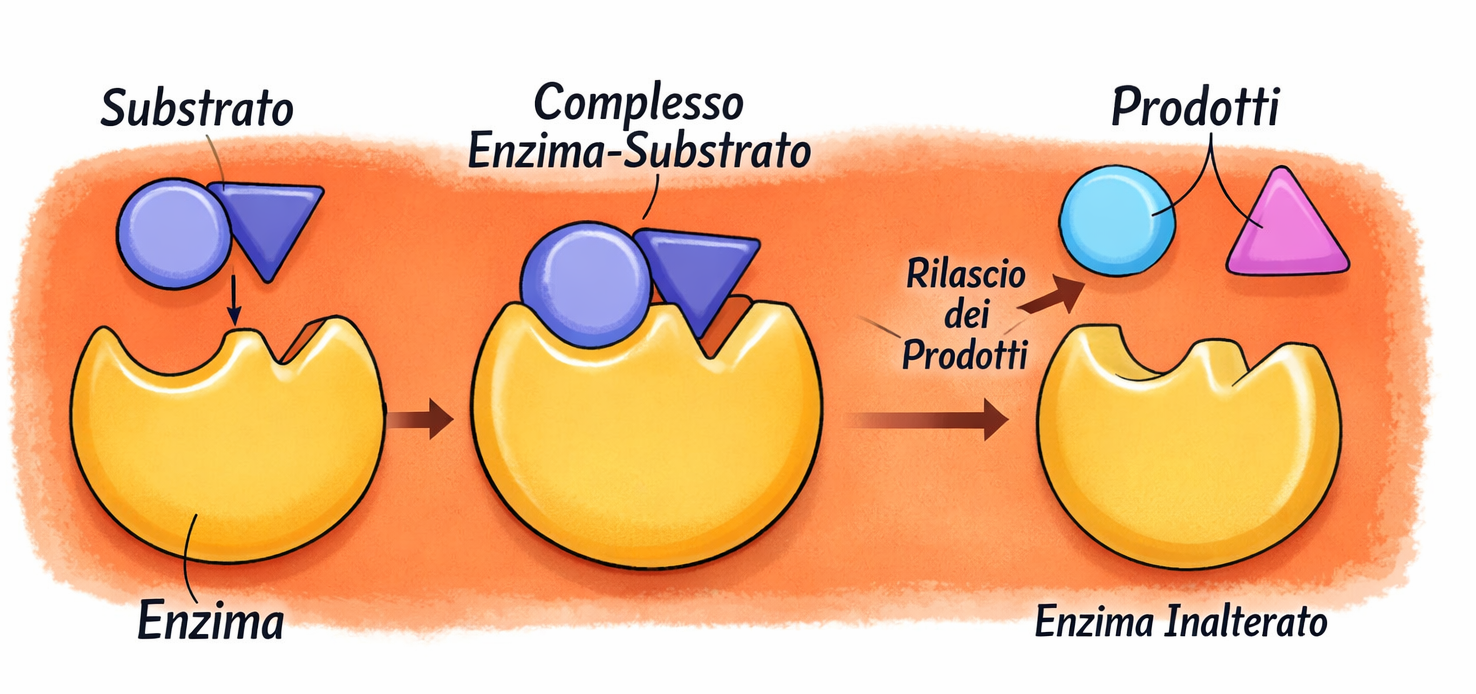
Image Gallery
Image Gallery
Molte molecole a uso farmacologico esercitano la loro efficacia bloccando selettivamente l’attività di enzimi bersaglio. Un esempio è rappresentato dalle statine, inibitori competitivi dell’HMG-CoA reduttasi, un enzima chiave della biosintesi del colesterolo a livello epatico; riducendo l’attività dell’enzima, diminuisce la produzione endogena di colesterolo. Il metotrexato, analogo dell’acido folico, inibisce la diidrofolato reduttasi e limita la disponibilità di tetraidrofolato necessario alla sintesi de novo dei nucleotidi, arrestando la proliferazione di cellule neoplastiche particolarmente dipendenti da un’elevata velocità replicativa. Nel campo dell’oncologia mirata, l’imatinib (Gleevec®) è stato progettato per occupare il sito di legame dell’ATP della chinasi tirosinica BCR-ABL, la cui attività aberrante promuove la leucemia mieloide cronica, impedendone la fosforilazione dei substrati.
Dal punto di vista meccanicistico, gli inibitori possono essere competitivi (aumentano il \(K_M\) apparente senza modificare \(V_{\max}\) poiché competono con il substrato per il sito attivo), non competitivi o misti (riduzione di \(V_{\max}\) per effetto sulla catalisi a prescindere dal legame del substrato), o irreversibili quando formano legami covalenti stabili con l’enzima. La scoperta di nuovi candidati spesso inizia con saggi ad alta processività su librerie di composti, seguiti da ottimizzazione chimica per incrementare affinità, selettività e proprietà farmacocinetiche del ligando.
Benché la sequenza amminoacidica determini struttura e attività delle proteine, molte funzioni biologiche richiedono il contributo di cofattori non proteici, indispensabili per realizzare trasformazioni chimiche non accessibili ai soli amminoacidi. Tali elementi includono ioni metallici, coenzimi organici e gruppi prostetici stabili. In termini didattici, è utile distinguere tra:
- cofattori metallici, come Zn\(^{2+}\), Fe\(^{2+}\)/Fe\(^{3+}\), Mg\(^{2+}\), coordinati da residui del sito attivo e cruciali per la catalisi o la stabilità;
- coenzimi organici, spesso derivati vitaminici, che partecipano come portatori attivati a trasferimenti di gruppi chimici o di equivalenti riducenti;
- gruppi prostetici, componenti organici strettamente e persistentemente associati alla proteina, talora tramite legami covalenti.
Esempi paradigmatici includono la rodopsina, recettore fotosensibile nei bastoncelli retinici, che sfrutta il retinale legato covalentemente a una lisina per assorbire fotoni; l’isomerizzazione fotoindotta del retinale innesca una cascata di trasduzione del segnale culminante in un impulso elettrico (Figura 01.17-09). L’emoglobina, d’altra parte, incorpora quattro gruppi eme, anelli porfirinici con un atomo di ferro centrale responsabile del legame reversibile con l’ossigeno molecolare e della colorazione rossa del sangue (Figura 01.17-11); (Figura 01.17-09).
Anche molti enzimi richiedono cofattori per svolgere la catalisi. La carbossipeptidasi lega in modo stretto uno ione zinco nel sito attivo: lo Zn\(^{2+}\) polarizza il gruppo carbonilico del substrato peptidico e stabilizza stati di transizione durante l’idrolisi. In altri sistemi, una piccola molecola organica agisce da trasportatore transitorio: la biotina, ad esempio, è un coenzima delle carbossilasi che trasferiscono gruppi carbossilici \((\text{–COO}^-)\), formando un legame covalente temporaneo con il gruppo trasferito e operando come “braccio mobile” tra siti attivi distinti (Figura 01.17-06). Poiché l’organismo umano non sintetizza la biotina in quantità adeguate, essa va introdotta con la dieta ed è perciò classificata come vitamina. Analogamente, la vitamina A alimentare è il precursore del retinale necessario alla funzione visiva.
Image Gallery

Image Gallery

Image Gallery
Image Gallery


