


Per studiare le cellule i biologi si servono di microscopi e strumenti di indagine biochimica
TOPICS
Definizione

La straordinaria varietà degli esseri viventi e la loro capacità di occupare ambienti estremamente diversi derivano dall’accumulo graduale di variazioni genetiche nel corso di milioni di anni. Tra queste, solo una frazione limitata risulta vantaggiosa, perché consente un migliore adattamento a condizioni ambientali in cambiamento; molte altre sono neutre o addirittura dannose. Nel breve periodo, e dal punto di vista del singolo organismo, è tuttavia indispensabile che il patrimonio genetico rimanga stabile: sopravvivenza e riproduzione richiedono un genoma affidabile. Tale stabilità è garantita non solo dall’elevata accuratezza dei sistemi di replicazione del DNA, ma anche da complessi apparati proteici che ispezionano continuamente il genoma per riconoscere e correggere eventuali lesioni. Sebbene una parte dei cambiamenti derivi da rari errori di copia, la maggioranza dei danni al DNA origina dalla chimica ordinaria della cellula, in particolare da reazioni spontanee e da metaboliti reattivi. Nella grande parte dei casi, il danneggiamento del DNA è transitorio, perché intervengono tempestivamente processi di correzione, collettivamente indicati come riparazione del DNA. L’importanza della riparazione è evidente quando tali vie falliscono. Negli individui affetti da xeroderma pigmentoso, per esempio, mutazioni ereditarie in geni chiave del sistema di riparazione impediscono la correzione dei danni indotti dalla radiazione ultravioletta (UV). L’accumulo di lesioni in cellule cutanee esposte alla luce solare favorisce la comparsa di gravi alterazioni tissutali, inclusi tumori. In modo analogo, difetti in altri meccanismi di controllo e riparazione sono associati a predisposizioni neoplastiche e a instabilità genomica. La fedeltà combinata di replicazione e riparazione risulta quindi determinante nel modellare l’integrità e l’evoluzione del nostro genoma.
Come tutte le macromolecole biologiche, il DNA è immerso in un ambiente dinamico, in cui urti termici e reazioni chimiche spontanee possono modificarne la struttura. Tra le alterazioni più comuni figurano:
- idrolisi spontanea dei legami glicosidici nelle purine (A e G), con formazione di siti abasici: la depurinazione elimina la base lasciando intatta l’ossatura fosfodiesterica, creando una “lacuna” simile a un dente mancante (Figura 03.03-01) e (Figura 03.03-03);
- deamminazione della citosina, che la converte in uracile, introducendo una base non fisiologica nel DNA e predisponendo a errori di appaiamento in replicazione (Figura 03.03-01);
- danni fotoindotti dalla radiazione UV, in particolare dimeri ciclobutanici tra pirimidine adiacenti come le timine, i quali distorcono l’elica e ostacolano l’avanzamento della replicazione (Figura 03.03-02);
- modifiche ossidative e alchilazioni dovute a specie reattive dell’ossigeno e ad altri metaboliti cellulari, che producono basi anomale e rotture nella catena;
- lesioni iatrogene o ambientali, causate da agenti chimici genotossici e da radiazioni ionizzanti, con possibili rotture a singolo o doppio filamento.
Le stime sperimentali indicano che, in una singola cellula umana, si generano ogni giorno decine di migliaia di lesioni di vario tipo; valutato su scala dell’intero organismo, ciò corrisponde a un numero complessivo di eventi estremamente elevato, fino a circa 5 × 10^16 depurinazioni nell’arco di 24 ore. Se non fossero prontamente riparate, tali alterazioni si tradurrebbero in conseguenze diverse: sostituzioni di coppie di basi per appaiamenti errati durante la replicazione (Figura 03.03-03), delezioni puntiformi per salto replicativo su siti abasici (Figura 03.03-03), o arresto del complesso replicativo in corrispondenza di lesioni ingombranti come i dimeri di timina. Oltre ai danni chimici, una quota di errori nasce dalla replicazione stessa. Nonostante l’elevata accuratezza del macchinario replicativo e l’attività di correzione di bozze, può occasionalmente essere incorporato un nucleotide scorretto che sfugge al proofreading. Per ciascuna di queste evenienze, la cellula mette in campo una o più vie di riparazione dedicate, in grado di rilevare la lesione, rimuoverla e ripristinare l’informazione corretta. L’incapacità di rimuovere dimeri di timina, come accade nello xeroderma pigmentoso, esemplifica come una singola classe di danno, se non gestita, determini accumulo di mutazioni e predisposizione al cancro. Danno e riparazione costituiscono dunque un equilibrio dinamico, essenziale per preservare la stabilità genomica.
Image Gallery

Image Gallery

Image Gallery
Image Gallery
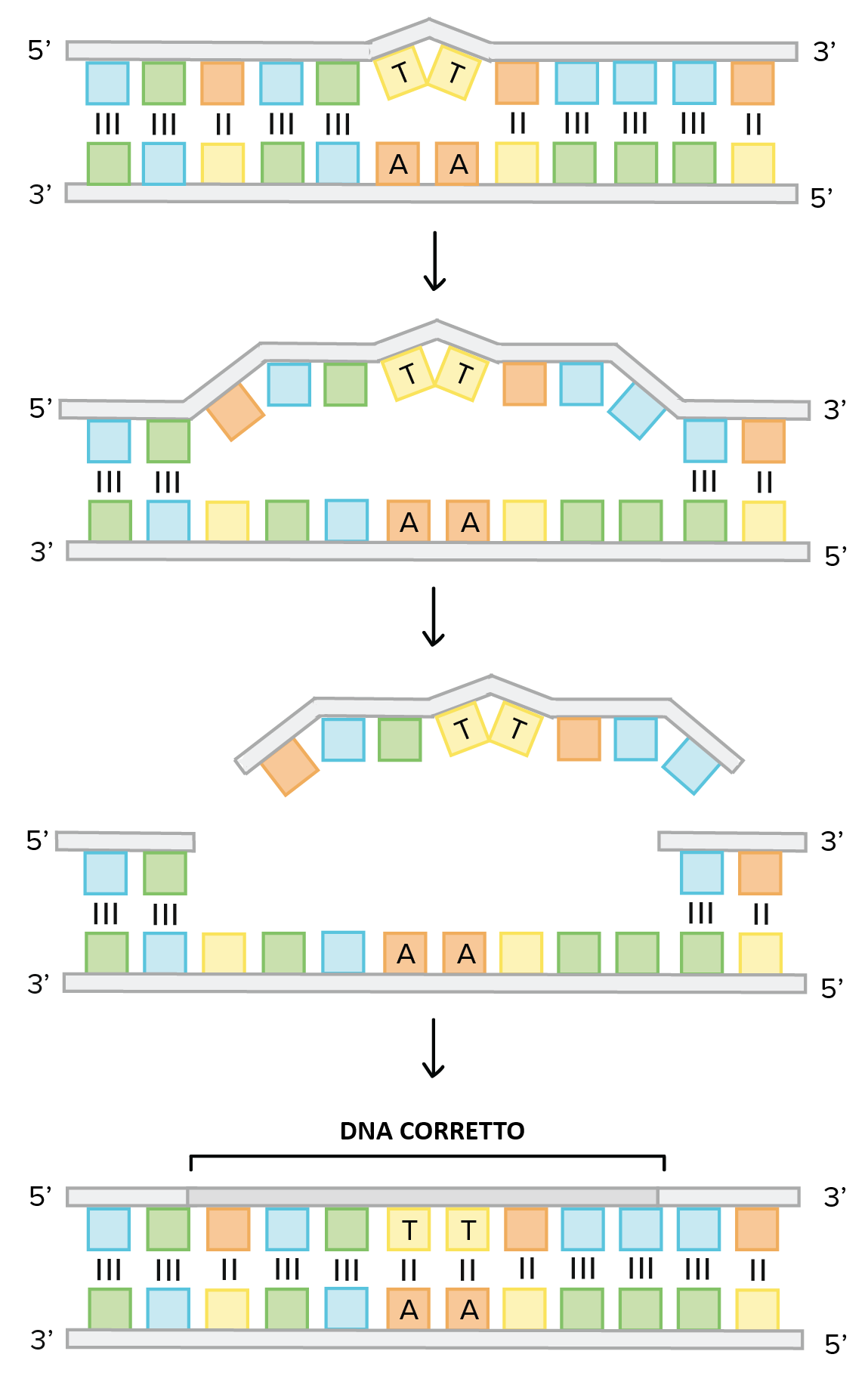
Image Gallery
Image Gallery
Le migliaia di alterazioni che insorgono quotidianamente nel DNA per effetto di idrolisi, metaboliti reattivi, agenti chimici e radiazioni vengono corrette da un repertorio coordinato di sistemi enzimatici. Il principio di fondo sfrutta la natura a doppio filamento del DNA: l’altra elica offre, nella maggior parte dei casi, una matrice integra da cui ricostruire l’informazione persa. Le strutture anomale introdotte dalle lesioni permettono al sistema cellulare di distinguere il filamento danneggiato da quello indenne. La via generale di ripristino, schematizzata in (Figura 03.03-04), può essere riassunta nelle seguenti fasi:
- riconoscimento e incisione: proteine sentinella individuano la distorsione o la base anomala e una nucleasi effettua un taglio mirato, rimuovendo la porzione danneggiata e lasciando un’interruzione su un solo filamento;
- risintesi a partire dal filamento sano: una DNA polimerasi di riparazione si associa all’estremità 3′ ossidrilica e colma il gap copiando l’informazione complementare; come le polimerasi replicative, agisce in direzione 5′→3′ ed è dotata di proofreading ad alta fedeltà; in molte cellule questo enzima coincide con quello che riempie i vuoti generati dalla rimozione degli inneschi di RNA durante la replicazione canonica;
- sigillatura: la DNA ligasi ricuce la discontinuità dello scheletro zucchero-fosfato, ristabilendo la continuità del filamento, analogamente a quanto avviene nell’unione dei frammenti di Okazaki del filamento lento.
A questa cornice comune si affiancano vie specializzate, selezionate in base alla natura del danno:
- riparazione per escissione di base (BER): riconosce basi anomale o siti abasici derivanti da deamminazione, ossidazione o depurinazione, mediante DNA glicosilasi, AP endonucleasi e successiva risintesi;
- riparazione per escissione di nucleotidi (NER): elimina ingombri elicoidali estesi, come i dimeri pirimidinici indotti dall’UV, tramite rimozione di un oligonucleotide contenente la lesione e ricostruzione del tratto;
- riparazione dei mismatches (MMR): corregge appaiamenti errati sfuggiti al proofreading replicativo, migliorando ulteriormente la fedeltà di copia;
- riparazione delle rotture a doppio filamento: ricorso a giunzione non omologa (NHEJ) o ricombinazione omologa (HR), a seconda della fase del ciclo cellulare e della disponibilità di un cromatide integro come stampo;
- polimerasi di translesione: in situazioni di blocco replicativo, polimerasi speciali bypassano temporaneamente la lesione, a costo di minore accuratezza, per poi demandare la correzione ai sistemi di escissione.
La concertazione tra riconoscimento del danno, rimozione, risintesi e ligazione, insieme ai controlli del ciclo cellulare e ai checkpoint che modulano replicazione e trascrizione in presenza di lesioni, salvaguarda l’integrità del genoma. Laddove questi meccanismi siano difettosi, l’instabilità genomica aumenta e con essa il rischio di malattie degenerative e oncologiche. La riparazione del DNA rappresenta quindi un pilastro della biologia cellulare, in equilibrio costante con le forze che generano danno e con l’inevitabile errore residuo della replicazione.
Image Gallery

Pur essendo estremamente accurato e dotato di attività di proofreading, l’apparato replicativo non è infallibile. Una quota minima di basi inserite in modo scorretto sfugge al controllo della polimerasi e dell’esonucleasi \(3' \rightarrow 5'\). In questi casi interviene un sistema di “secondo livello”, la riparazione degli appaiamenti errati del DNA (DNA mismatch repair, MMR), che sorveglia e corregge i mismatch residui. In media, il complesso replicativo introduce circa un errore ogni \(10^7\) nucleotidi incorporati; l’azione del MMR rimuove la stragrande maggioranza di questi difetti (oltre il 99%), portando il tasso complessivo intorno a un errore ogni \(10^9\) nucleotidi copiati, una precisione di gran lunga superiore a quella dei processi tecnologici ordinari (Tabella 03.03-01). Se un mismatch permane fino al ciclo replicativo successivo, esso si fissa come mutazione stabile (Figura 03.03-05).
Il MMR riconosce e corregge coppie di basi non complementari e piccoli loop derivanti da scivolamenti della polimerasi, in particolare nelle regioni ripetute. Il complesso di riparazione identificata la discrepanza, incide e rimuove un tratto di uno dei due filamenti contenente l’errore, quindi ricostruisce il segmento mancante mediante una DNA polimerasi e sigilla l’interruzione con una ligasi, ripristinando la sequenza corretta (Figura 03.03-06). Un passaggio cruciale è capire quale dei due filamenti rappresenti il “nuovo” contenente l’errore: sostituire il filamento stampo consoliderebbe la mutazione.
La discriminazione del filamento con l’errore avviene in modi diversi nei vari domini della vita. Nei batteri, la metilazione emiemimetilata delle sequenze GATC da parte di Dam consente di distinguere per un breve intervallo il DNA parentale (già metilato) da quello appena sintetizzato (non ancora metilato). Negli eucarioti, la selettività si basa su segnali strutturali e di processo: i nick fisiologici degli Okazaki fragment nella catena ritardata, la direzionalità del clamp PCNA e incisioni endonucleasiche guidate dal complesso MutLα indirizzano l’escissione verso il filamento neosintetizzato. Le proteine di riconoscimento includono MutSα (MSH2–MSH6) e MutSβ (MSH2–MSH3) per il rilevamento di mismatch e loop, mentre MutLα (MLH1–PMS2) coordina incisione ed escissione; exonucleasi e fattori di rimodellamento della cromatina completano la rimozione, seguiti da risintesi e ligazione.
Il ruolo del MMR nella prevenzione tumorale è ben documentato. Mutazioni germinali in geni del MMR causano predisposizione ereditaria a neoplasie, in particolare il carcinoma del colon-retto associato a instabilità dei microsatelliti (Lynch syndrome). Poiché i due alleli vengono ereditati separatamente, un individuo portatore di una sola variante patogenetica può rimanere asintomatico finché, in una cellula somatica, non si perde o inattiva anche l’allele sano. La progenie di quella cellula, priva del MMR, accumula mutazioni a ritmo accelerato e presenta un rischio elevato di trasformazione neoplastica. Tale dinamica clonale spiega perché difetti nei correttori di appaiamento determinino un marcato incremento del rischio oncologico.
Tassi di errore nella replicazione del DNA
| Situazione | Frequenza di errore |
|---|---|
| Dattilografo a 120 parole/minuto | 1 errore ogni 250 caratteri |
| Trasporto bagagli in aereo | 1 valigia persa o danneggiata ogni 400 passeggeri |
| Incidenti automobilistici negli Stati Uniti | 1 decesso ogni 10⁴ persone all’anno |
| Replicazione del DNA senza proofreading | 1 errore ogni 10⁵ nucleotidi copiati |
| Replicazione del DNA con proofreading ma senza correzione mismatch | 1 errore ogni 10⁷ nucleotidi copiati |
| Replicazione del DNA con tutte le correzioni attive | 1 errore ogni 10⁹ nucleotidi copiati |
| Tabella che descrive i principali meccanismi di correzione degli errori durante la replicazione del DNA (proofreading e riparazione), responsabili della drastica riduzione della frequenza di errore e della stabilità genetica cellulare. | |
Image Gallery

Image Gallery

Le vie di riparazione considerate finora sfruttano la ridondanza informativa intrinseca alla doppia elica: quando un filamento è danneggiato, quello complementare funge da stampo per il ripristino. Le rotture a doppio filamento (double-strand breaks, DSB), invece, pongono una sfida diversa, poiché interrompono simultaneamente entrambe le catene e possono condurre alla perdita o al riarrangiamento di porzioni cromosomiche. Fonti di DSB includono radiazioni ionizzanti, agenti che bloccano la forcella replicativa, specie reattive dell’ossigeno e stabilizzazione di complessi di topoisomerasi. In assenza di uno stampo immediatamente disponibile, la cellula rischia di non poter ricomporre fedelmente l’informazione.
Due sono le principali strategie evolute per gestire le DSB. La prima è la ricongiunzione non omologa delle estremità (non-homologous end joining, NHEJ), un processo rapido che ricollega le estremità rotte prima che si disperdano. Complessi comprendenti Ku70/Ku80 si legano ai capi del DNA, reclutando DNA-PKcs, nucleasi per la pulizia delle estremità (ad esempio Artemis), polimerasi specializzate e la ligasi IV con XRCC4–XLF, che sigillano la rottura. Questa via è efficiente ma intrinsecamente imprecisa: la “rifilatura” delle estremità e l’aggiunta non templata di nucleotidi possono eliminare o inserire basi, con rischio di mutazioni o piccole delezioni (Figura 03.03-07). Quando il danno coinvolge regioni codificanti o regolative, le conseguenze possono essere severe. Nonostante ciò, la NHEJ è essenziale, in particolare nelle fasi del ciclo in cui uno stampo omologo non è accessibile, e viene sfruttata fisiologicamente, ad esempio, nella ricombinazione V(D)J dei linfociti.
In alternativa, la cellula può ricorrere a una via ad alta fedeltà, la ricombinazione omologa (homologous recombination, HR), che utilizza una copia omologa intatta come stampo per ricostruire con precisione l’informazione genetica persa (Figura 03.03-07). Questa strategia è particolarmente attiva in fase S e G2, quando la cromatide sorella, appena replicata e trattenuta in prossimità dal complesso coesina, è disponibile come riferimento.
Image Gallery
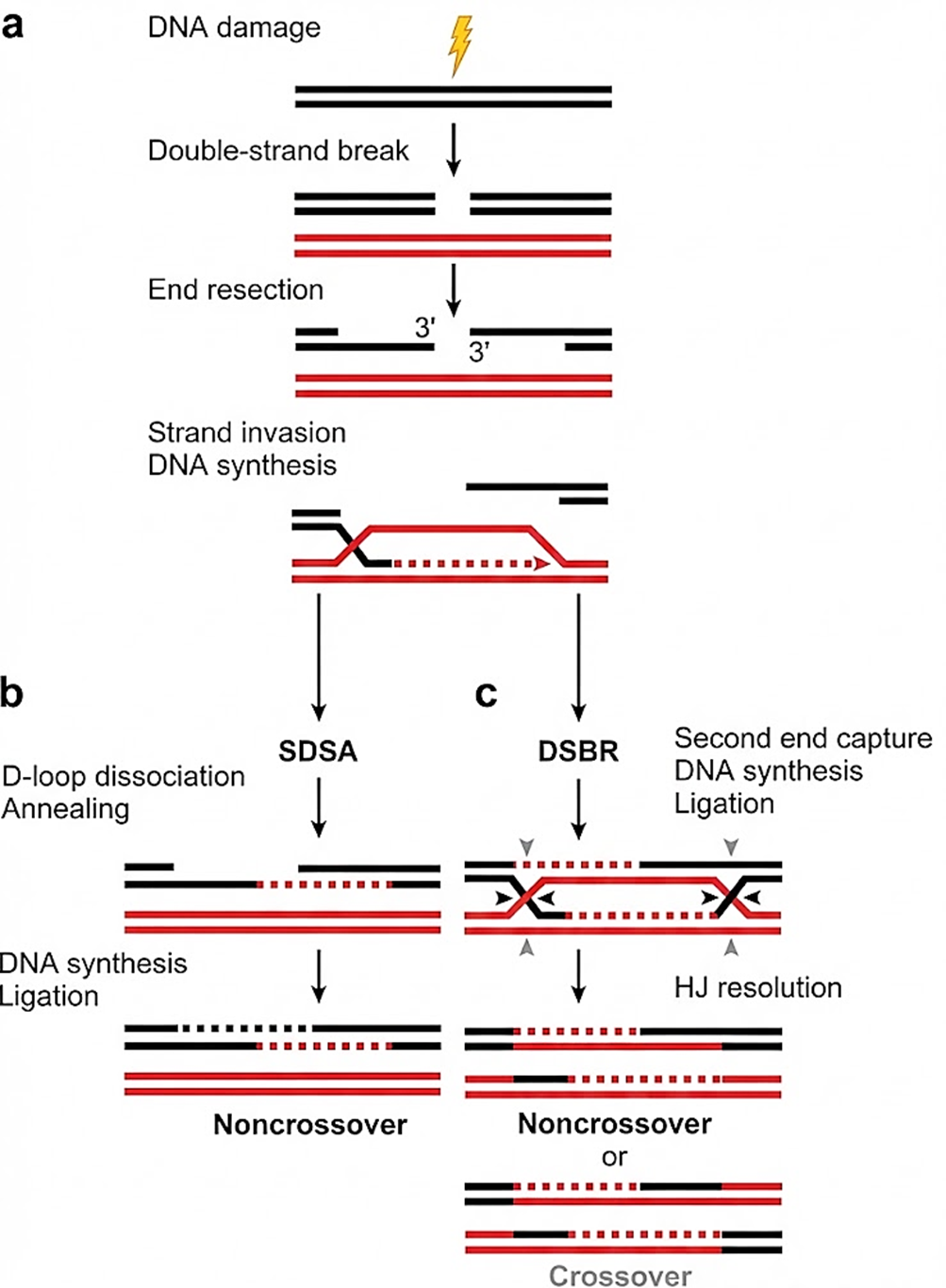
Image Gallery
La sfida principale nella riparazione di una DSB è reperire una matrice intatta. Subito dopo la replicazione di un tratto di DNA, la cromatide sorella costituisce una copia omologa ideale per guidare il ripristino: le sequenze identiche su entrambi i lati della rottura permettono un appaiamento accurato e una ricostruzione senza perdita di informazioni. In queste condizioni, la HR procede in più passaggi coordinati (Figura 03.03-08) e può raggiungere una riparazione “a prova d’errore”.
Il processo generalmente si avvia con una resezione delle estremità rotte: il complesso MRN (MRE11–RAD50–NBS1), insieme a CtIP, rimuove nucleotidi dalle estremità \(5'\) generando code a singolo filamento \(3'\) (Figura 03.03-08). Le regioni a singolo filamento vengono inizialmente coperte da RPA, quindi sostituite da filamenti di Rad51 (omologo eucariotico di RecA) con l’assistenza di mediatori come BRCA2 e, in lievito, Rad52. Il nucleoprotein filament guidato da Rad51 cerca una sequenza omologa e promuove l’“invasione” della doppia elica intatta, formando un D-loop e appaiandosi estensivamente con il filamento complementare (Figura 03.03-08). Una DNA polimerasi di riparazione estende l’estremità \(3'\) del filamento invasore utilizzando come stampo la cromatide sorella (Figura 03.03-08).
Una volta superato il punto di rottura, il filamento neo-sintetizzato può rientrare nella molecola originaria e riappaiarsi con il proprio complementare (Figura 03.03-08). Il completamento avviene mediante ulteriore sintesi a partire dalle estremità \(3'\) di entrambi i filamenti interrotti (Figura 03.03-08), seguito da ligazione (Figura 03.03-08). Varianti del percorso, quali la “sintesi-dipendente dall’appaiamento del filamento” (SDSA) o la formazione di giunzioni di Holliday doppie, vengono poi risolte da specifiche endonucleasi o dissolte da complessi elicasi-topoisomerasi (ad esempio BLM–TopoIIIα–RMI). Il risultato sono due doppie eliche integre, con informazione ripristinata fedelmente grazie allo stampo omologo.
La HR è altamente conservata in tutti i domini della vita, riflettendo la sua versatilità: interviene non solo nelle DSB ma anche nel recupero di forcelle replicative collassate e nella riparazione di lesioni complesse. Inoltre, durante la meiosi, la ricombinazione omologa mediatizza lo scambio di tratti cromosomici tra omologhi, contribuendo in modo determinante alla variabilità genetica delle popolazioni.
Image Gallery
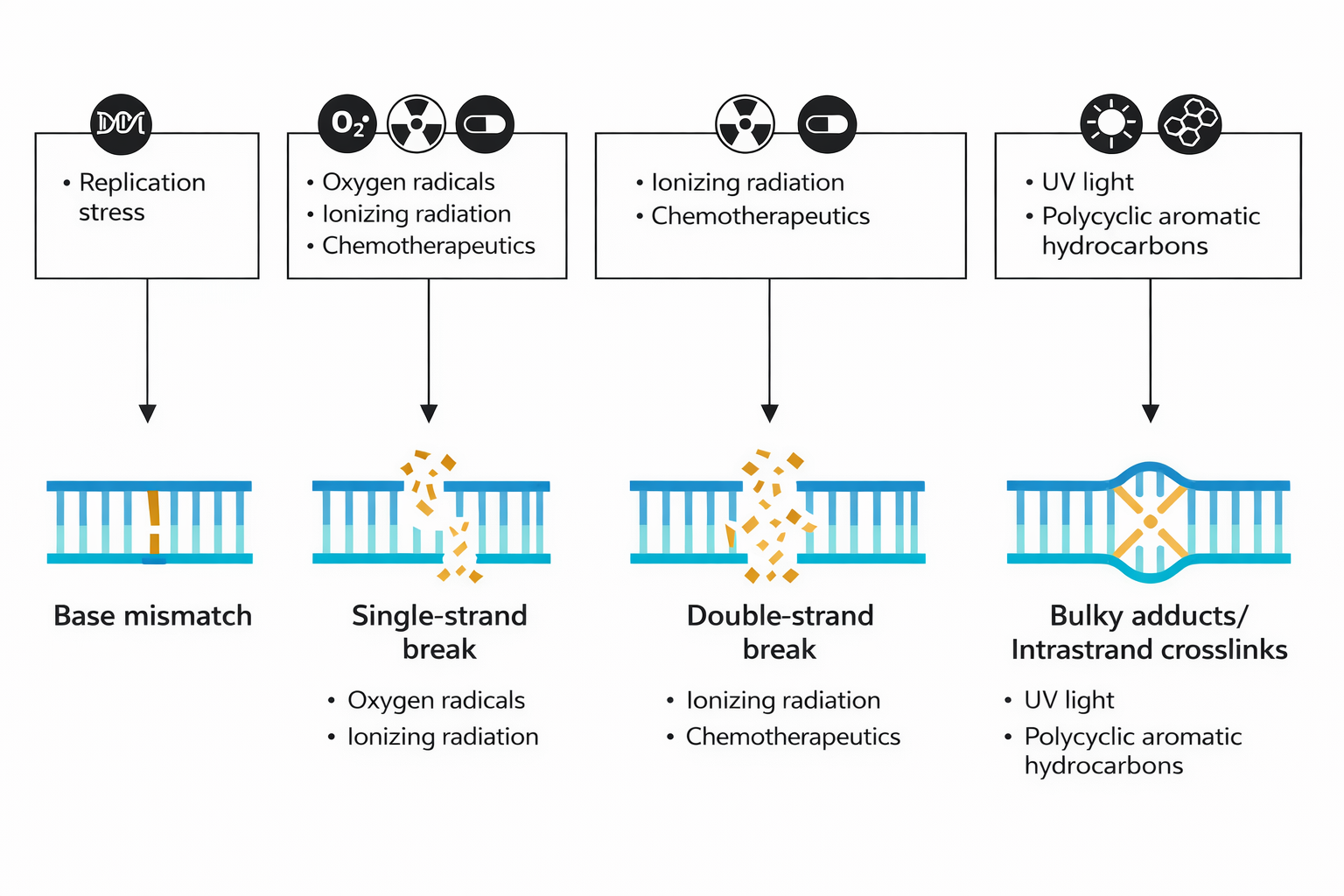
Image Gallery
Image Gallery
Image Gallery
Image Gallery
Image Gallery
Image Gallery
Gli errori non corretti nati durante replicazione o riparazione si trasformano in mutazioni permanenti. Se la mutazione modifica la sequenza di una proteina in modo da alterarne stabilità, conformazione o interazioni, la funzione cellulare può risentirne in maniera marcata. Un singolo cambiamento di base nel gene HBB, che sostituisce l’acido glutammico con valina in posizione 6 della catena β, produce l’emoglobina S responsabile dell’anemia falciforme. In condizioni di bassa tensione di ossigeno, l’emoglobina S tende a polimerizzare, irrigidendo gli eritrociti in forme allungate e fragili (Figura 03.03-09); ne derivano emolisi, anemia e possibili occlusioni microvascolari con dolore e danno d’organo. Curiosamente, l’eterozigosi per la variante S conferisce una protezione parziale contro la malaria, illustrando come la selezione naturale possa mantenere alleli svantaggiosi in specifici contesti ambientali.
Anche nelle cellule somatiche l’accumulo progressivo di alterazioni puntiformi, delezioni, inserzioni e riarrangiamenti può generare cloni con vantaggi proliferativi. Il cancro rappresenta l’esito estremo di questa evoluzione clonale: una sequenza di mutazioni casuali, favorita da difetti nei sistemi di riparazione o da esposizioni mutagene, conduce a crescita incontrollata, evasione dei segnali di arresto e instabilità genomica (Figura 03.03-10). Un modesto incremento del tasso mutazionale a livello di popolazione cellulare, ad esempio per perdita del MMR o per deficit nella HR, può moltiplicare la probabilità di acquisire combinazioni di lesioni oncogeniche e accelerare l’insorgenza tumorale. Considerando che circa il 30% dei decessi in Europa e Nord America è imputabile a patologie neoplastiche, l’elevata fedeltà della duplicazione e dei circuiti di riparazione del DNA è cruciale tanto per le linee germinali, che trasmettono l’informazione alle generazioni successive, quanto per i tessuti somatici, in cui l’omeostasi dipende da un controllo genetico stringente:
- difetti nel MMR determinano instabilità dei microsatelliti, facilitando l’accumulo di frameshift in geni con tratti ripetuti, inclusi regolatori del ciclo cellulare;
- compromissioni della HR, ad esempio mutazioni in BRCA1/2, rendono le cellule vulnerabili a DSB non riparate e conferiscono sensibilità a inibitori di PARP per letalità sintetica;
- l’eccessivo ricorso alla NHEJ su DSB multiple può favorire traslocazioni e aneuploidie, contribuendo all’instabilità cromosomica propria di molte neoplasie.
Accanto all’anemia falciforme, mutazioni puntiformi in altri geni possono produrre fenotipi clinici severi: per esempio, una sostituzione attivante nel recettore FGFR3 è alla base della acondroplasia, mentre varianti loss-of-function in PAH causano fenilchetonuria se non trattate con restrizione dietetica. Questi esempi sottolineano come anche minimi cambiamenti della sequenza possano avere ricadute funzionali sostanziali.
Image Gallery
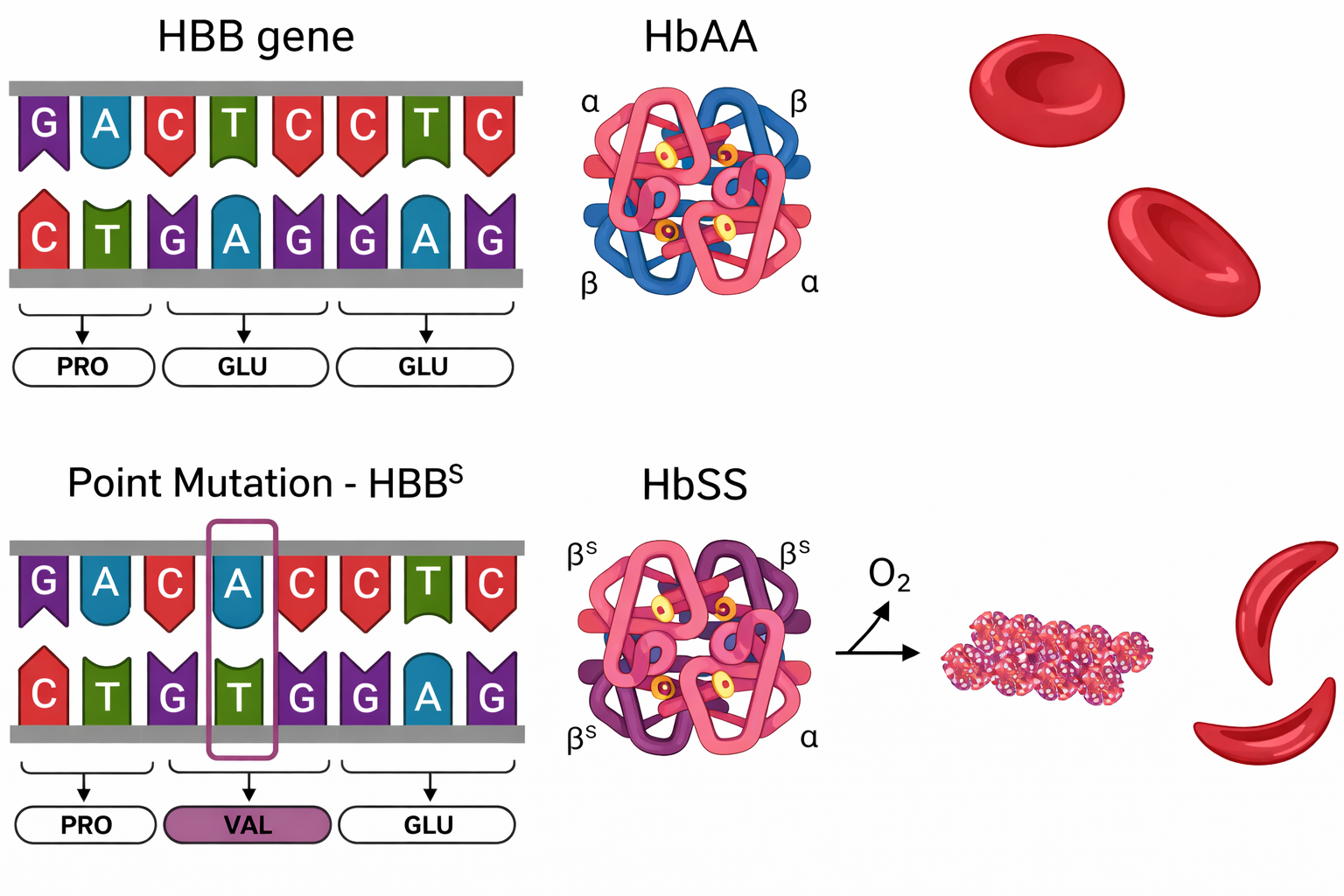
Image Gallery
Le mutazioni deleterie tendono a essere eliminate dalla selezione naturale, poiché riducono la sopravvivenza o la capacità riproduttiva degli organismi; quelle vantaggiose possono fissarsi e diffondersi. Tuttavia, anche nei siti neutri, dove la variazione nucleotidica non incide sensibilmente sulla fitness, l’accuratezza dei processi di copia e riparazione fa sì che il messaggio ereditario si conservi a lungo. La parentela stretta tra esseri umani e scimpanzé, separatisi evolutivamente pochi milioni di anni fa, si riflette in un’elevata omologia di sequenza, con identità che supera il 98% su ampie porzioni del genoma. Anche confronti fra specie più distanti, come essere umano e balena, mostrano corrispondenze cromosomiche e blocchi sintenici chiaramente riconoscibili, nonostante tempi di divergenza di ordine 10–20 volte maggiori (Figura 03.03-11). In altri termini, i genomi conservano una traccia storica in cui solo una piccola frazione del contenuto informativo essenziale è stata alterata lungo centinaia di milioni di anni. La combinazione di alta fedeltà replicativa, efficaci sistemi di riparazione e pressione selettiva ha preservato l’architettura e il significato funzionale di gran parte della sequenza, consentendo al contempo la variabilità necessaria all’evoluzione.
Image Gallery



