Trasduzione del segnale: una sequenza di interazioni molecolari trasferisce i segnali dai recettori alle molecole relè nella cellula
Definizione

Le librerie genomiche e di cDNA rimangono strumenti essenziali per clonare geni molto estesi e per il sequenziamento su larga scala; tuttavia, per numerose applicazioni il ricorso alla reazione a catena della polimerasi (PCR, Polymerase Chain Reaction) ha reso l’amplificazione e il clonaggio di regioni definite di DNA più rapide e operative in provetta. Introdotta alla fine del XX secolo, la PCR ha trasformato l’analisi degli acidi nucleici consentendo la replicazione mirata e selettiva di un segmento specifico di DNA, con un incremento esponenziale del numero di copie nel giro di un’ora circa, senza l’uso di cellule batteriche. La PCR sfrutta la complementarietà delle basi per dirigere una DNA polimerasi verso la regione bersaglio definita da due primer sintetici. Il requisito principale è la conoscenza, almeno parziale, della sequenza del bersaglio per progettare i primer; grazie ai repertori genomici oggi disponibili in banche dati pubbliche, tale vincolo è raramente limitante. L’alta sensibilità della PCR permette di rivelare quantità minime di DNA, ad esempio tracce in un campione ambientale o clinico, rendendola utile oltre il clonaggio: dalla diagnostica molecolare alla sorveglianza epidemiologica fino alla medicina forense.
La PCR si fonda su due proprietà: la specificità di ibridazione dei primer al loro stampo e la capacità di una DNA polimerasi termostabile di estendere un filamento a partire da un’estremità 3′-OH. I primer, brevi oligonucleotidi a singolo filamento, definiscono i confini dell’amplicone e forniscono l’estremità 3′ necessaria all’enzima per iniziare la sintesi. Progettati sulla base della sequenza bersaglio e sintetizzati chimicamente, i primer fungono sia da inneschi sia da “indirizzi” che guidano l’enzima verso la porzione di DNA da amplificare. La reazione procede per cicli ripetuti di tre fasi, realizzati in un termociclatore:
- denaturazione, separazione dei due filamenti del DNA stampo a temperatura elevata, tipicamente ~94–98 °C;
- appaiamento dei primer (annealing), con ibridazione dei primer alle regioni complementari a una temperatura dipendente dalla loro temperatura di melting, in genere ~50–65 °C;
- estensione, durante la quale la DNA polimerasi sintetizza nuovo DNA a partire dai primer, spesso a ~72 °C per enzimi derivati da Thermus aquaticus.
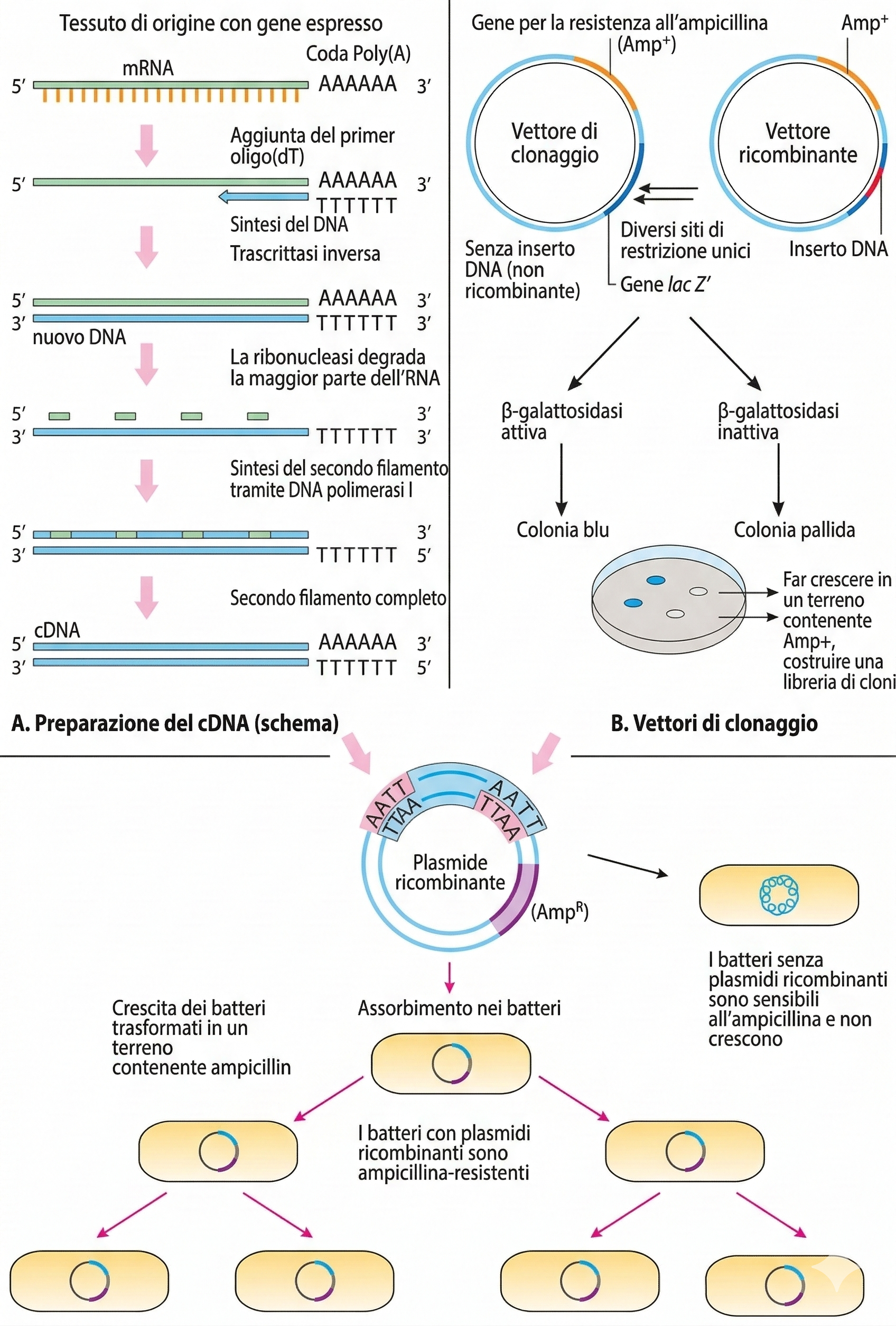
All’inizio di ogni ciclo i filamenti del DNA stampo vengono separati e ciascun primer si appaia alla sequenza complementare; la polimerasi poi estende da ciascun primer in direzione 5′→3′ (Figura 04.08-01). Le molecole neo-sintetizzate diventano a loro volta stampi per i cicli successivi, determinando un aumento esponenziale delle copie (Figura 04.08-02). In condizioni ideali, dopo n cicli il numero teorico di molecole è descritto da \( N = N_0 \cdot 2^{n} \), dove \(N_0\) è il numero iniziale di copie. Per esempio, partendo da 30 copie e dopo 25 cicli, il numero atteso, in assenza di inefficienze, è \(30 \cdot 2^{25}\), pari a oltre 1,0 \times 10^9 copie. La scelta della polimerasi è cruciale: Taq DNA polimerasi offre rapidità ma non possiede attività correttiva 3′→5′, mentre enzimi ad alta fedeltà (per esempio Pfu o Phusion) riducono gli errori grazie al proofreading, risultando preferibili quando il prodotto deve essere clonato ed espresso. Parametri come concentrazione di Mg2+, purezza dei dNTP, temperatura e durata di annealing/estensione influenzano selettività ed efficienza; design dei primer (18–25 nucleotidi, contenuto GC ~40–60 %, assenza di dimeri e hairpin stabili) e numero di cicli modulano la qualità dell’amplificazione. La PCR è particolarmente adatta a frammenti inferiori a 10 000 coppie di basi; approcci “long-range” con polimerasi ottimizzate possono estendere questo limite, ma con maggiore attenzione alle condizioni di reazione. Lo stampo può essere DNA genomico o plasmidico; quando il punto di partenza è RNA, una trascrittasi inversa converte l’mRNA in cDNA, che diventa il substrato per la PCR, consentendo di ottenere l’amplicone privo di introni (Figura 04.08-03). Un vantaggio sostanziale è la possibilità di generare direttamente un frammento clonabile a partire da campioni complessi senza costruire previamente una libreria. Per facilitare l’inserimento in vettori, i primer possono includere al 5′ siti di restrizione o sequenze di ricombinazione; inoltre, la natura delle estremità dipende dall’enzima impiegato (ad esempio, Taq aggiunge frequentemente un’adenina in 3′, utile per il “TA cloning”). Per ridurre falsi positivi dovuti a contaminazioni, si adottano buone pratiche di laboratorio e, quando opportuno, sistemi con dUTP/uracil-DNA glicosilasi per degradare carry-over di prodotti precedenti. Questi accorgimenti preservano la specificità che è alla base dell’efficacia della tecnica.
Image Gallery

Image Gallery

Image Gallery
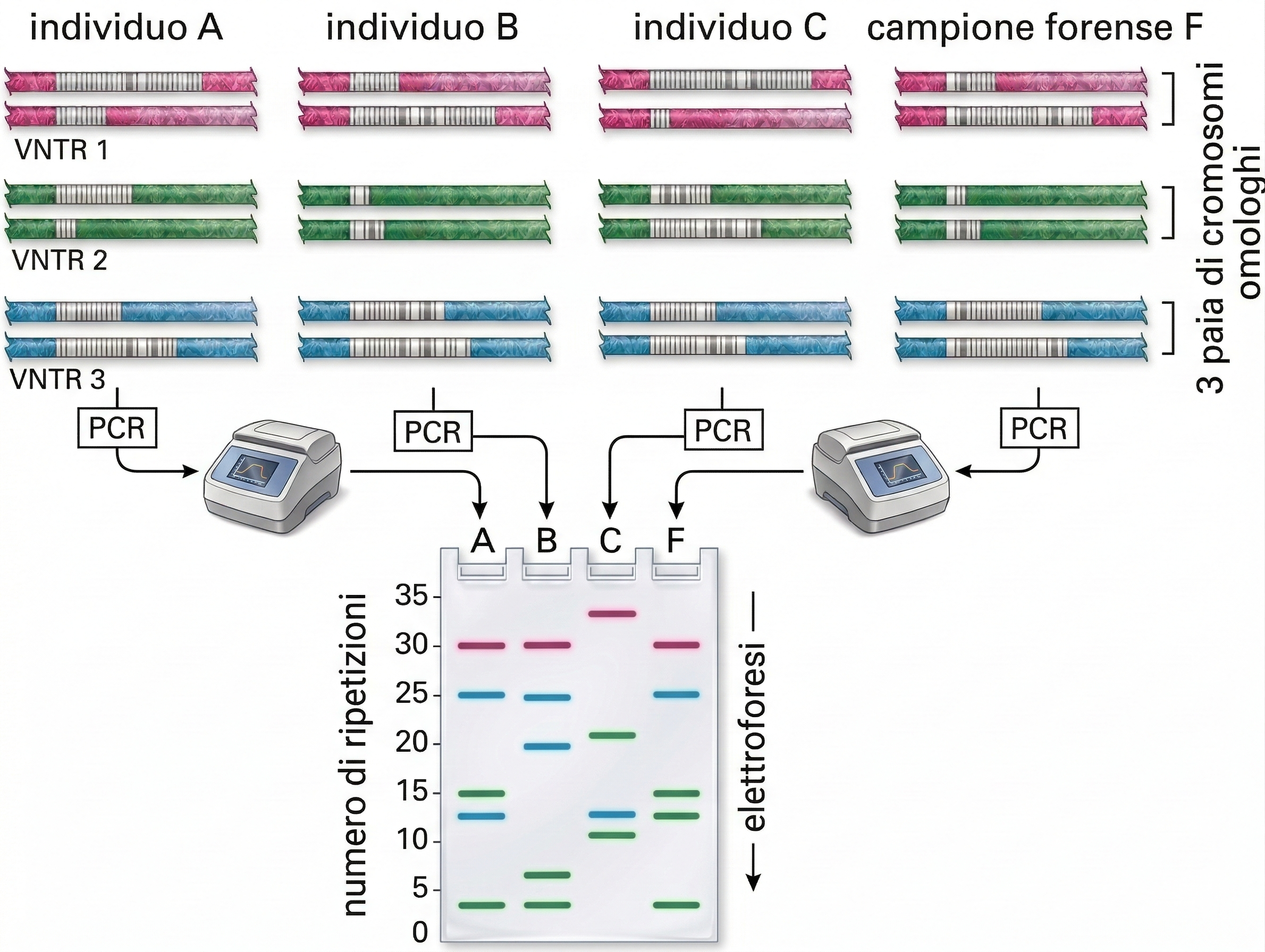
Oltre al clonaggio, la PCR è ampiamente impiegata per l’identificazione rapida di microrganismi patogeni, anche quando il carico infettivo è molto basso. Primer specifici per regioni del genoma di batteri, virus o parassiti consentono di rivelare poche copie virali o batteriche in campioni clinici dopo molteplici cicli di amplificazione (Figura 04.08-04). Varianti quantitative della tecnica, come la PCR in tempo reale, permettono di stimare la carica microbica e di monitorare la risposta a una terapia. La stessa logica sostiene attività di sanità pubblica e sicurezza: la PCR contribuisce a tracciare focolai epidemici, a riconoscere precocemente agenti coinvolti in eventi bioterroristici e a controllare prodotti alimentari per la presenza di microrganismi o di componenti non dichiarate. È inoltre possibile verificare l’autenticità di ingredienti, per esempio confermando l’origine animale o vegetale di una materia prima in alimenti lavorati, o identificando adulterazioni. In ambito forense, la straordinaria sensibilità della PCR consente la genotipizzazione a partire da quantità minime di materiale biologico, quali un singolo bulbo pilifero o cellule epiteliali su un oggetto. L’analisi di loci altamente polimorfici, tipicamente brevi ripetizioni in tandem (STR), mediante multiplex PCR produce un profilo unico della persona cui appartiene il DNA, esclusi i gemelli monozigoti (Figura 04.08-05). L’informazione così ottenuta è utilizzabile per associare campioni a individui, per escludere sospettati o per identificare vittime, nel rispetto di rigorosi standard di qualità e catena di custodia. Per garantire l’affidabilità dei risultati in diagnostica e forense, si impiegano controlli di processo (controlli negativi e positivi), standard di riferimento e protocolli validati. La combinazione di sensibilità, rapidità e specificità rende la PCR uno strumento di prima scelta quando è necessario amplificare e caratterizzare sequenze definite di DNA con precisione e tempestività.
Image Gallery
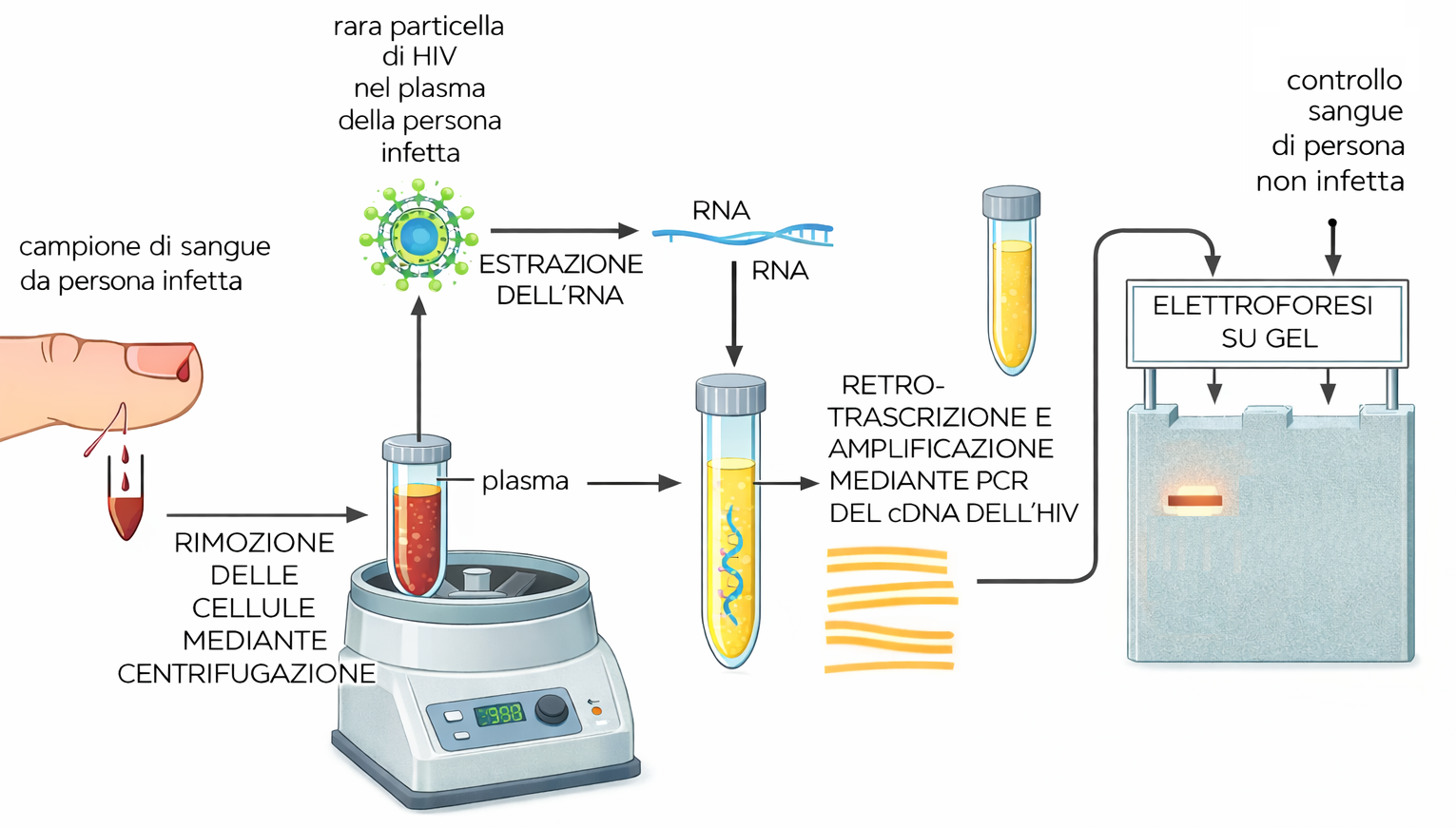
Image Gallery